الموجز
أمراض تخزين الدهون السفينغولية (Sphingolipidoses) هي مجموعة من الاضطرابات
الأيضية الموروثة التي تتميّز بتراكم
الدهون (الشحميّات) السفينغولية في الأنسجة المختلفة؛ بسبب نقصٍ أو خللٍ في
إنزيماتليسوسومية تساهم في
أيض هذه الدهون. تعتبر هذه الأمراض من اضطرابات الاختزان في
الليسوسومات، وتظهر مجموعة واسعة من الأعراض السريرية على المرضى، مثل: التدهور العصبي، وتضخّم الأعضاء، والتشوهات الجلدية. تشمل أمراض تخزين
الدهون السفينغولية البارزة مرض غوشيه (Gaucher’s disease)، ومرض فابري (Fabry disease)، ومرض تاي ساكس (Tay-Sachs disease)، ومرض نيمان بيك (Niemann-pick disease)، ومرض كرابي (Krabbe disease)، وحثل المادة البيضاء المتبدل اللون (Sulfatide lipidosis, Metachromatic leukodystrophy)، ويرتبط كل منها بنقصٍ أو خللٍ في
إنزيم معيّن، ومن ثم تراكُم
دهون سفينغولية محددة. يتضمن تشخيص هذه الأمراض اختبارات بيوكيميائية لقياس نشاط
الإنزيمات، واختبارات جينية لتحديد
الطفرات المسببة لها، ودراسات تصويرية لتقييم الأنسجة المصابة. تركّز استراتيجيات العلاج على العلاج التعويضي للإنزيمات، والعلاج بتخفيض الركيزة (Substrate)،
والعلاج الجيني، وزرع
الخلايا الجذعية المكوّنة للدم، والرعاية الداعمة لإدارة الأعراض وتحسين نوعية الحياة.
تعريفها
أمراض تخزين
الدهون السفينغولية (سفينغوليبيدوزس، Sphingolipidoses) هي مجموعة من الاضطرابات
الأيضية الموروثة التي تتميز بالتراكم غير الطبيعي للدهون السفينغولية داخل
الأجسام الحالّة (الليسوسومات، Lysosomes). تتراكم
الدهون غير المتحللة نتيجةً لنقص في واحد أو أكثر من
الإنزيمات الليسوسومية المحللة لهذه
الدهون. يكون النقص في
الإنزيمات في العادة وراثيًا، فلا ينتج
الإنزيم، أو يتكوّن بصورة خاطئة، أو قد يتكون ولكن فعاليته تكون ضعيفة أو معدومة. ونتيجة عدم قدرة
الليسوسوم على هدم بعض
الدهون السفينغولية بشكل صحيح، تتكدس
الدهون هناك، وتؤدي إلى مجموعة من الأعراض السريرية التي تختلف اعتمادًا على أي
إنزيم قد نقص، وعلى نوع الدهن السفينغولي المتراكم، وغيرها من الاعتبارات الأخرى[1].
تُعدّ أمراض تخزين
الدهون السفينغولية مجموعة فرعية من
اضطرابات الاختزان في الليسوسومات (Lysosomal storage disorders)، وهي فئة أوسع من الأمراض
الأيضية الوراثية الناتجة من خلل في وظائف
الإنزيمات الليسوسومية[2]. وتشمل اضطرابات الاختزان أكثر من 50 اضطرابًا مختلفًا، وكلها تنطوي على مشاكل في تكسير الجزيئات الكبيرة وتراكمٍ لاحق لهذه الجزيئات المختلفة داخل الأجسام الحالّة؛ ما يسبّب تعطيل الوظيفة الطبيعية
للخلية، ويؤدي إلى سلسلة من الأحداث الخلوية المتتابعة المسبِّبة للمشاكل المختلفة، مثل الالتهاب (Inflammation) والإجهاد التأكسدي (Oxidative stress) والموت الخلوي المبرمج. كل هذه المشاكل الخلوية تؤدّي لاحقًا إلى أعراض سريرية وأمراض مختلفة للكائن الحي. إن دراسة أمراض تخزين
الدهون السفينغولية في سياق اضطرابات الاختزان في
الليسوسومات أمر ضروري، إذ إن الآلية المشتركة لاختلال الوظيفة
الليسوسومية تساعد في فهم الأسس الجزيئية لهذه الأمراض، وتساهم في تطوير علاجات موجّهة تهدف إلى استعادة وظيفة
الليسوسومات وتخفيف الآثار الضارّة لهذه الاضطرابات[3].
أهمية الدهون السفينغولية في الوظائف الخلوية
الدهون أو
الشحميّات السفينغولية، وتُعرف أيضًا بالسفينغوليبيدات (Sphingolipids)، جزيئات بيولوجية كبيرة ومعقدة (Macromolecules) تندرج تحت قسم الشحوم. والدهون السفينغولية، مثل
الدهون الفسفورية (Phospholipids) والستيرولات (Sterols)، هي مكوّنات أساسية لأغشية الخلايا، وتساهم في سلامة البنية الهيكلية ووظيفة
الأغشية والخلايا. لا يقتصر دور
الدهون السفينغولية على هيكلية
الخلايا، بل تشارك في العديد من العمليات الخلوية، بما في ذلك نقل الإشارات (Signal transduction)، والتواصل بين
الخلايا (Cell-cell interaction)، وتنظيم عمليات
شيخوخة الخلايا {{شيخوخة الخلايا: (Cell senescence) حالة خلوية تتوقف فيها دورة حياة الخلية، ولكنها لا تموت. تحدث هذه الحالة استجابةً لمجموعة من الضغوط الداخلية والخارجية على الخلية مثل تلف الحمض النووي، أو تقصير التيلوميرات، أو الإجهاد التأكسدي، أو النشاط السرطاني. تفيد شيخوخة الخلايا في التئام الجروح والتطور الجنيني، ولكن تراكمها يساهم في شيخوخة الكائن الحي وكثرة أمراض التليّف وتصلّب الشرايين والتنكس العصبي.}} والاستماتة الخلوية (موت الخلايا المبرمج، Apoptosis). تعمل الكثير من
الدهون السفينغولية بصفتها جزيئات
دهنية نشطة بيولوجيًا (Bioactive lipids) تؤثر في مسارات الإشارات الخلوية التي تتحكّم في نمو
الخلايا وتمايزها (Cell differentiation) وبقائها. كما تجمع بعض
الدهون السفينغولية بين الوظيفة الهيكلية والوظيفة البيولوجية، فتتشارك مع الكولسترول في تكوين الطوافات الدهنية (lipid rafts)، وهي حيّزات غشائية صغيرة (Membrane microdomains) متخصصة تتشكّل في الغشاء الخلوي وتسهّل تنظيم
البروتينات لتكوين مركّبات نقل الإشارة (Signaling complexes)، ومن ثَم تحفّز وظائف هذه المركّبات. بالإضافة إلى كل ذلك، للدهون السفينغولية أدوار أساسية في الإدخال الخلوي (Endocytosis)، والإخراج الخلوي (Exocytosis)، والتنقّل في داخل
الخلايا (Intracellular trafficking)؛ ما يسهّل الاتصالات بين
الخلايا وداخلها. إن مركّبات نقل الإشارة تساهم أيضًا في الحفاظ على التوازن الخلوي وصحة
الخلايا، فهي تُنظّم عملية التمثيل الغذائي للدهون السفينغولية إلى حد كبير، كما هو الحال مع معظم العمليات
الأيضية الخلوية. لذلك، فإن الاختلال في المسارات الاستقلابية للدهون السفينغولية داخل
الخلية يؤدي إلى حالات مرضية شديدة.
الخلفية التاريخية
يعود تاريخ اكتشاف أمراض تخزين
الدهون السفينغولية إلى أواخر القرن التاسع عشر وأوائل القرن العشرين، عندما بدأ العلماء والأطباء بتحديد الاضطرابات
الأيضية الموروثة التي تتميز بتراكم غير طبيعي للدهون في الأنسجة ووصفها. شكّلت هذه الاكتشافات بداية لفهم أمراض التخزين في الجسيمات الحالّة، وأرست الأساس للبحوث البيوكيميائية والوراثية الحديثة في هذه المجالات. كان أول مرض يوصف من هذه العائلة هو مرض غوشيه، الذي اكتشفه الطبيب الفرنسي فيليب إرنست غوشيه (Philippe Ernest Gaucher، 1854-1918) في عام 1882، فقد أبلغ عن حالة مريض يعاني من تضخّم الطحال، ولاحظ وجود
خلايا كبيرة محمّلة بالدهون في الطحال[4]. وكان عملُ غوشيه رائدًا في التعرف على المرض الجديد، مع أن الخلل البيوكيميائي الأساسي لم يُكتَشَف إلا بعد ذلك بكثير بوساطة العالم روسكو بريدي (1923-2016) عام 1965[5].
كان المرض الثاني الذي اكتُشف هو تاي ساكس الذي وصفه عالمان بشكل مستقل بعضهما عن بعض وسُمّي على اسميهما. ففي عام 1881 لاحظ طبيب العيون البريطاني وارن تاي (Warren Tay، 1843-1927) البقعة الحمراء الكرزية المميزة على شبكية العين في ثلاث حالات من نفس العائلة اليهودية الشرقية[6]. أما طبيب الأعصاب الأميركي برنارد ساكس (Bernard Sachs، 1858-1944)، فوصف عام 1887 التنكّس العصبي التدريجي وتوقّف نمو
الدماغ الذي يُرى في الأطفال المصابين[7]. أصبح الارتباط بين البقعة الحمراء الكرزية والتدهور العصبي علامة مميزة للمرض، إذ اكتشف الباحثان شينتارو أوكادا وجون أوبراين (John S. O'Brien)أنه ناتج من نقص في
إنزيم بيتا هيكسوزأمينيداز أ (β-hexosaminidase A) عام 1969[8].
أما مرض فابري، فوصفه طبيب الجلد الألماني يوهانس فابري (Johannes Fabry، 1860-1930) والجرّاح البريطاني ويليام أندرسون (William Anderson، 1842-1900) عام 1898، حين عَرَضا، بشكل مستقل، حالات لمرضى مصابين بتقرّحات جلدية وآلام في الأطراف واختلالات في وظائف الكلى[9]. ولم يكن يُعرف حتى عام1952، حين عُرف أن سبب المرض تراكمٌ غير طبيعي في
الدهون. وفي عام 1970، اقترن المرض بنقص
الإنزيم ألفا غالاكتوسيداز أ[10]؛ ما أدّى إلى فهم أعمق لتطوّر المرض. أما مرض حثل المادة البيضاء المتبدل اللون، فقد وصفه لأول مرة عالم الأعصاب والطبيب النفسي الألماني ألويس آلزهايمر (Alois Alzheimer، 1864-1915) وتلميذه الإيطالي غيتانو بيروسيني (Gaetano Perusini, 1879-1915) عام 1910 في المرضى البالغين[11]. وفي عام 1925، نشر عالم الأعصاب الألماني ڤيليبالد أوسكار شولتز (Willibald Oscar Scholz، 1889-1971) دراسة سريرية عن هذا المرض عند الأطفال[12]. فحص فايفر الأنسجة العصبية في دراسة شولتز عام 1959 ووجد أنها كانت تحتوي على أقسام مصبوغة بألوان متعددة[13]. وبشكل مستقل اكتشف هورست ياتزكيفيتز (Horst Jatzkewitz، 1912-2002) أن الأقسام الملطخة بالصبغة نتجت من تراكم السولفاتيدات[14]. ومن ناحية أخرى، اكتشف جيمس أوستن، عام 1963، أن الخلل في نشاط
إنزيم آريل سولفاتاز أ (Arylsulfatase A) هو المسبب لهذا المرض[15].
في أوائل القرن العشرين، وتحديدًا عام 1914، وصف الطبيب الألماني ألبرت نيمان (Albert Niemann، 1834-1921) ما يُعرف الآن بمرض نيمان بيك النوع أ (Niemann-Pick type A) لأول مرة، وأكمل وصفَ حالاته المرضية عالمُ الأمراض الألماني لودفيغ بيك (Ludwig Pick، 1868-1944) في ثلاثينيات القرن العشرين. وصف ألبرت نيمان حالةً مصابةً بتضخم الكبد والطحال وأعراض عصبية أخرى[16]، في حين تضمّن عمل بيك دراسات نسيجية كشفت عن وجود
خلايا رغوية مليئة بترسبات دهنية في الأعضاء المصابة[17]. جاء اكتشاف نقص
إنزيم السفينغومياليناز الحمضي (Acid sphingomyelinase) بوصفه مسببًا للمرض في وقت لاحق[18]. أما في عام 1916، فوصف طبيب الأعصاب الدنماركي كنود كرابي (Knud Krabbe، 1885-1961) مرض كرابي، فحدّد المرض عند الرضّع الذين يعانون من أعراض عصبية شديدة ووصف وجود
خلايا كروية في المادة البيضاء في
الدماغ[19]. وفي سبعينيات القرن العشرين اكتُشف نقص
الإنزيم الأساسي غالاكتوسيريبروسيداز (Galactocerebrosidase) بصفته مسؤولًا أصيلًا عن هذا المرض[20].
شهد منتصف القرن العشرين تقدّمًا كبيرًا في التقنيات البيوكيميائية؛ ما أدّى إلى تحديد أوجه القصور
الإنزيمية المحدّدة المسؤولة عن أمراض تخزين
الدهون السفينغولية. وسمح ظهور علم الوراثة الجزيئي في الجزء الأخير من القرن العشرين باستنساخ
الجينات التي تشفّر هذه
الإنزيمات؛ ما سهّلَ الفحوصَ الجينية وتشخيص الأمراض قبل الولادة. في الأعوام الأخيرة من القرن العشرين، ركّزت الأبحاث على فهم الآليات الجزيئية التي تكمن وراء هذه العائلة من الأمراض لتطوير أساليب علاجية جديدة، بما في ذلك
العلاج الجيني والعلاج بتقليل الركيزة[21].
تصنيفها
تُصنّف أمراض تخزين
الدهون السفينغولية على أساس نقص
الإنزيمالليسوسومي المحدّد الذي يؤدي إلى تراكم أنواع مختلفة من
الدهون السفينغولية. يعرض (الجدول 1) أمراض تخزين
الدهون السفينغولية الموروثة المعروفة والإنزيم المختلّ ونوع ركيزة الدهن السفينغولي التي تتراكم في
الخلايا. يتميز كل من هذه الأمراض بخصائص مرضية وسريرية مختلفة. يساعد هذا التصنيف في فهم الفيزيولوجيا المرضية والمظاهر السريرية وطرق العلاج المحتملة لكل اضطراب[22].
[الجدول 1]
تصنيف أمراض تخزين الدهون السفينغولية المختلفة
المرض | الإنزيم المعتلّ[23]
| النواتج الرئيسة المتراكمة[24] | علاجه |
|---|
مرض غوشيه (Gaucher disease) | غلوكوسيريبروسيداز (Glucocerebrosidase) | غلوكوزيل سيراميد (Glucosylceramide) | ممكن بوساطة العلاج الإنزيمي التعويضي (Enzyme replacement therapy) |
مرض فابري (Fabry disease) | ألفا-غالاكتوسيداز (α-galactosidase) |
الدهون السفينغولية السكرية (Glycosphingolipids) | ممكن بوساطة العلاج الإنزيمي التعويضي |
مرض تاي ساكس (Tay-Sachs disease) | بيتا هيكسوزأمينيداز أ (β-hexosaminidase A) | غانغليوسيد جي إم 2 (GM2 ganglioside) | غير متوفر |
مرض نيمان بيك (Niemann-Pick disease) | سفينغومياليناز (Sphingomyelinase) |
سفينغوميالين (Sphingomyelin) | غير متوفر |
مرض كرابي (Krabbe disease) | غالاكتوسيريبروسيداز (Galactocerebrosidase) | غالاكتوزيل سيراميد (Galactosylceramide) | ممكن بواسطة زراعة نخاع العظم (Bone marrow transplant) |
حثل المادة البيضاء متبدل اللون (Metachromatic leukodystrophy) | آريل سولفاتاز أ (Arylsulfatase A) | سولفاتيد (Sulfatide) | ممكن بوساطة زراعة نخاع العظم |
جي إم 1 غانغليوسيدوسز (GM1 gangliosidosis) | بيتا-غالاكتوسيداز (β -galactosidase) | غانغليوسيد جي إم 1 (GM1 ganglioside) | غير متوفر |
مرض ساندهوف (Sandhoff disease) | بيتا هيكسوزأمينيداز أ وب (β-hexosaminidase A and B) | غانغليوسيد جي إم 2 (GM2 ganglioside) وغانغليوسيد جي أيه 2 (GA2 ganglioside) | غير متوفر |
مرض فاربر (Farber disease) | سيراميداز (Ceramidase) |
سيراميد | غير متوفر |
مرض غوشيه
يحدث مرض غوشيه(Gaucher’s Disease) نتيجة نقص في
إنزيم غلوكوسيريبروسيداز المسؤول عن تكسير غلوكوزيل سيراميد إلى
سيراميد وغلوكوز {{الغلوكوز: (C₆H₁₂O₆) سكر أحادي سداسي الكربون، وهو المصدر الرئيس للطاقة لمعظم الكائنات الحية، والناتج الرئيس لعملية التمثيل الضوئي (Photosynthesis)، والركيزة الأساسية للتنفس الخلوي (Cellular respiration). يدور في الدم على شكل سكر الدم، ويُخزن على شكل غلايكوجين (Glycogen) في الكبد والعضلات لدى الحيوانات أو على شكل نشا (Starch) في النباتات. يتحلل في الخلية لإنتاج طاقة بوساطة التحلل الغلايكولي والفسفرة التأكسدية (Oxidative phosphorylation).}} (الشكل 1). عندما لا يعمل هذا
الإنزيم بالصورة المطلوبة، تتراكم جزيئات الغلوكوزيل
سيراميد وغيرها من الغلوكوسيريبروسيدات في
الليسوسومات {{الليسوسومات: عضيّات خلوية حمضية تحتوي على إنزيمات حالّة لجميع الجزيئات الخلوية الكبيرة المعقدة.}}؛ ما يؤدي إلى اعتلالات مرضية في الطحال والكبد ونخاع العظام، في المقام الأول[25]. هناك ثلاثة أنواع من مرض غوشيه: النوع الأول (غير العصبي)، والنوع الثاني (الاعتلال العصبي الحاد)، والنوع الثالث (الاعتلال العصبي المزمن). النوع الأول هو الأكثر شيوعًا والأقل خطورةً، وتكون أعراضه الأساسية متمثلّة بتضخم الكبد والطحال وفقر الدم وآلام العظام، ومن الممكن أن تتأثر الرئتان والكلى في بعض الأحيان. ينطوي النوعان الثاني والثالث، وهما أشد خطورة، على أعراض عصبية شديدة، إذ يتطور النوع الثاني بسرعة ويؤدي إلى الوفاة في مرحلة الطفولة، بينما يتطور النوع الثالث بشكل أبطأ، وفي النوعين الثاني والثالث هناك أيضًا تضخم للكبد والطحال كما النوع الأول[26].
[الشكل 1]
/_تفاعل-إنزيم-غلوكوسيريبروسيداز-يزيل-مركب-الغلوكوز-من-مركب-غلوكوسيل-سيراميد-بمساعدة-بروتين-سابوزين-ج-.png) تفاعل إنزيم غلوكوسيريبروسيداز يزيل مركب الغلوكوز من مركب غلوكوسيل سيراميد بمساعدة بروتين سابوزين ج
تفاعل إنزيم غلوكوسيريبروسيداز يزيل مركب الغلوكوز من مركب غلوكوسيل سيراميد بمساعدة بروتين سابوزين ج
حذف الصورة؟
سيؤدي هذا إلى نقل الصورة إلى سلة المهملات.
يعتبر النوع الثاني أخطر أنواع المرض ولا يوجد علاج حالي له، بسبب تطوره السريع الذي يؤدي إلى الوفاة في عمر الطفولة، ولكن هناك علاجات ممكنة للمرضى الذين يعانون من النوع الأول ولجزء كبير ممن يعانون من النوع الثالث. إن العلاج الإنزيمي التعويضي باستخدام
الإنزيم الناقص (الغلوكوسيريبروسيداز) في الوريد يمكن أن يقلّل من تضخّم الكبد والطحال، ويقلل من تطور المرض، ولكن هذا العلاج مكلف للغاية، إذ إن التكلفة على المريض الواحد تتجاوز مئتي ألف دولار أميركي سنويًا، ويجب الاستمرار فيه مدى الحياة[27]. هناك أيضًا أدوية واعدة قد تساعد في تقليل أعراض مرض غوشيه من النوع الأول، مثل الميغلوستات (Miglustat) والإيليغلوستات (Eliglustat). دواء الميغلوستات الذي اعتُمد في أوروبا عام 2002 بصفته علاجًا لمرض غوشيه يُعطى عن طريق الفم ويمنع تكوين الغلوكوسيريبروسيد. وفي عام 2014، اعتُمِد الإيليغلوستات الذي يعمل عن طريق تثبيط إنتاج الغلوكوزيل سيراميد. يعتبر الدواءان جزءًا من نهج علاجي يسمى العلاج بتقليل الركيزة (Substrate reduction therapy)[28].
مرض فابري
ينتج مرض فابري(Fabry Disease) من نقص في
إنزيم ألفا غالاكتوسيداز أ[29]؛ ما يؤدي إلى تراكم
الدهون السفينغولية السكرية، وخصوصًا غلوبو تري أوسيل سيراميد، جي بي 3 (Globotriaosylceramide, Gb3) في الأنسجة المختلفة (الشكل 2). يؤثر هذا الاضطراب المرتبط بالكروموسوم X عند الذكور بشكل أكثر شدة منه عند الإناث، مع أعراض تشمل الألم في الأطراف (تنميل الأطراف، Acroparesthesia) والأورام الوعائية القرنية (Angiokeratomas)، ونقص التعرق (Hypohidrosis)، وعتامة القرنية (Corneal opacity). ومع تقدم المرض، يمكن أن يؤدي إلى مضاعفات تهدّد الحياة مثل الفشل الكلوي (Renal failure)، وتضخّم القلب (Cardiac hypertrophy)، وغيرها من المشاكل
الدماغية[30].
[الشكل 2]
 وفيه يُزال جزيء سكر مكونًا لاكتوزيل سيراميد/_تفاعل-إنزيم-ألفا-غالاكتوسيداز-أ-الذي-يتم-بمساعدة-بروتين-سابوزين-ب-(Saposin-B)-وفيه-يُزال-جزيء-سكر-مكونًا-لاكتوزيل-سيراميد.png) تفاعل إنزيم ألفا غالاكتوسيداز أ الذي يتم بمساعدة بروتين سابوزين ب (Saposin B) وفيه يُزال جزيء سكر مكونًا لاكتوزيل سيراميد
تفاعل إنزيم ألفا غالاكتوسيداز أ الذي يتم بمساعدة بروتين سابوزين ب (Saposin B) وفيه يُزال جزيء سكر مكونًا لاكتوزيل سيراميد
حذف الصورة؟
سيؤدي هذا إلى نقل الصورة إلى سلة المهملات.
ومثل مرض غوشيه، فإن علاج مرض فابري يكون بشكل أساسي عبر استخدام العلاج الإنزيمي التعويضي الباهظ التكلفة. من الجدير ذكره أن هذا العلاج ليس علاجًا شافيًا، ولكنه يؤخّر تطوّر المرض بطريقة جزئية، وقد يعكس بعض الأعراض السريرية. في هذا العلاج، يأخذ المريض جرعة وريدية مرة كل أسبوعين تتكون من
إنزيم ألفا غالاكتوسيداز تحت أسماء تجارية مختلفة، وهذا العلاج متاح في العديد من دول العالم، ولكن تكاليفه مرتفعة للغاية. من الممكن أيضًا استخدام استراتيجية علاج أخرى وهي العلاج بالمرافق الدوائي (Pharmacological chaperone therapy)، وفيها يُستخدَم المرافق الدوائي للحفاظ على نشاط
الإنزيم من خلال المساعدة في تكوين
البروتين المختلّ بشكل صحيح على الرغم من
الطفرات الموجودة فيه والمُسببة للمرض. حتى عام 2024، يتوفر دواء واحد يعتمد هذه الاستراتيجية وهو ميغالاستات (Migalastat) الذي يؤخذ عن طريق الفم[31]. بالإضافة إلى هذه الخيارات العلاجية، هناك أيضًا علاجات تجريبية دوائية لم يُوافَق عليها بعد.
مرض تاي ساكس
ينتج مرض تاي ساكس(Tay-Sachs Disease) من نقص في
إنزيم بيتا هيكسوزأمينيداز أ؛ ما يؤدي إلى تراكم
غانغليوسيد {{الغانغليوسيدات: دهون سفينغولية سكرية مُتَسَيْلِلَة (تحتوي على حمض السياليك).}} جي إم 2 في
الخلايا العصبية (الشكل 3). يتميز هذا الاضطراب الوراثي المتنحي (Autosomal recessive) بالتدهور السريع للجهاز العصبي المركزي. هناك عدة أشكال للمرض، والشكل الأكثر شيوعًا هو الطفولي، الذي يُظهِر تأخرًا في النمو وضعفًا في العضلات والبقعة الحمراء الكرزية المميزة على شبكية العين؛ ما يؤدي إلى ضعف عصبي شديد والوفاة في مرحلة الطفولة المبكرة. وهناك أيضًا أشكال مرضية تبدأ في فئة الشباب والبالغين بأعراض أخف. حتى نهاية عام 2024، لا يوجد علاج لأسباب مرض تاي ساكس ولا علاجات يمكن أن تبطئ تطوره.
[الشكل3]
 محوّلًا غانغليوسيد جي إم 2 إلى غانغليوسيد جي إم 3/_-----~3.PNG) تفاعل إنزيم بيتا هيكسوزأمينيداز أ الذي يساعده بروتين محفّز جي إم 2 (GM2 activator) محوّلًا غانغليوسايد جي إم 2 إلى غانغليوسايد جي إم 3
تفاعل إنزيم بيتا هيكسوزأمينيداز أ الذي يساعده بروتين محفّز جي إم 2 (GM2 activator) محوّلًا غانغليوسايد جي إم 2 إلى غانغليوسايد جي إم 3
حذف الصورة؟
سيؤدي هذا إلى نقل الصورة إلى سلة المهملات.
مرض نيمان بيك
بصورة عامة، ينتج مرض نيمان بيك(Niemann-Pick Disease) من نقص في
إنزيم السفينغومياليناز الحمضي (الشكل 4)؛ ما يؤدي إلى تراكم
السفينغوميالين. يؤدي تراكم
السفينغوميالين في
الجهاز العصبي المركزي إلى العديد من المشاكل العصبية، مثل: الخلل في الكلام، والصعوبة في البلع، والترنّح في المشي. ويؤدي المرض الأكثر شدة الذي يصيب القشرة المخية إلى فقدان القدرات الفكرية وتزايد النوبات التشنّجيّة. هناك ثلاثة أنواع من المرض: النوع أ وهو الشكل العصبي الحاد (Acute neuropathic) الذي يحدث في مرحلة الطفولة مع تدهور عصبي شديد ووفاة مبكرة، والنوع ب وهو الشكل غير العصبي (Non-neuropathic) الذي يظهر مع أعراض مثل تضخّم الكبد والطحال وأمراض الرئة وتشوهات العظام، ولكن مع متوسط عمر طبيعي. أما النوع ج من مرض نيمان بيك، فهو مختلف، وينتج بسبب عيوب في
بروتينات نقل
الكولسترول وليس في
إنزيم السفينغومياليناز كما النوعين السابقين[32]. لمرض نيمان بيك ج مجموعة واسعة من الأعراض العصبية. لا توجد، حتى نهاية عام 2024، أية أدوية معتمدة لعلاج هذا المرض، ولكن هناك العديد من الأدوية التجريبية قيد الدراسة، ويُنظر كذلك في
العلاج الجيني، إلا أنه لم يُوافق عليه بعد.
[الشكل4]
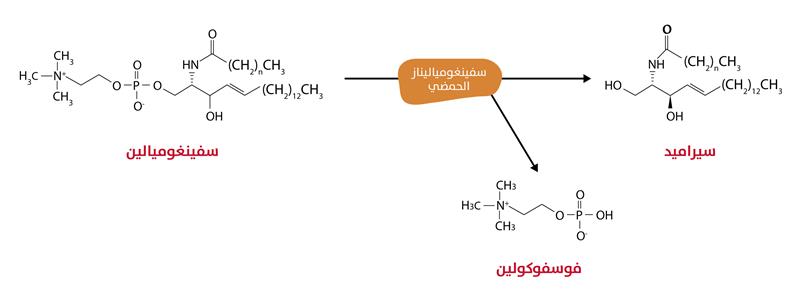 تفاعل إنزيم السفينغومياليناز
تفاعل إنزيم السفينغومياليناز
حذف الصورة؟
سيؤدي هذا إلى نقل الصورة إلى سلة المهملات.
مرض كرابي
ينتج مرض كرابي(Krabbe Disease)، المعروف أيضًا باسم حثل المادة البيضاء كروي الخلايا (Globoid cell leukodystrophy) وهو من عائلة أمراض حثل المادة البيضاء، بسبب نقص في
إنزيم غالاكتوسيريبروسيداز (الشكل 5)؛ ما يؤدي إلى تراكم غالاكتوزيل سيراميد المعروف أيضًا باسم سايكوسين (Psychosine)، وهو
دهن سفينغولي سام. يؤثر هذا الاضطراب المتنحي في المقام الأول في المادة البيضاء في
الدماغ والأعصاب الطرفية. تبدأ عوارض الشكل الطفولي من هذا المرض بصعوبات في التغذية وتأخر شديد في النمو، وتتطور إلى التشنجات، والنوبات العصبية، ومن ثم الوفاة عادةً بحلول سن الثانية. للمرض أيضًا أشكال متأخرة في الظهور بدرجات متفاوتة في الشدة. على الرغم من عدم وجود علاج معروف لمرض كرابي، فقد ثبت أن زراعة نخاع العظم أو زراعة
الخلايا الجذعية المكوّنة للدم قد تفيد بعض المرضى إذا تمّت في وقت مبكر من مسار المرض[33].
[الشكل5]
 وسابوزين ج (Saposin C) لكي ينتج السيراميد من غالاكتوزيل سيراميد/_-----~4.PNG) تفاعل إنزيم غالاكتوسيريبروسيداز الذي يحتاج إلى كل من سابوزين أ (Saposin A) وسابوزين ج (Saposin C) لكي ينتج السيراميد من غالاكتوزيل سيراميد
تفاعل إنزيم غالاكتوسيريبروسيداز الذي يحتاج إلى كل من سابوزين أ (Saposin A) وسابوزين ج (Saposin C) لكي ينتج السيراميد من غالاكتوزيل سيراميد
حذف الصورة؟
سيؤدي هذا إلى نقل الصورة إلى سلة المهملات.
مرض حثل المادة البيضاء المتبدل اللون
يحدث مرض حثل المادة البيضاء المتبدل اللون بسبب نقص في
إنزيم أريل سولفاتاز أ؛ ما يؤدي إلى تراكم السلفاتيدات، وخصوصًا السيريبروسايد سلفات (الشكل 6)، في
الجهاز العصبي وأعضاء أخرى. يؤدي هذا الاضطراب الجسدي المتنحي إلى إزالة
الميالين التدريجي من حول
الخلايا العصبية؛ ما يجعل الأعصاب عرضة لعدم العمل بصورة جيدة ومن ثم الموت. الشكل الطفولي المتأخر من المرض هو الأكثر شيوعًا، وتتمثل أعراضه باختلال الأعصاب الحركية والتراجع الحركي، والترنّح في الحركة (Ataxia)، والتدهور الإدراكي (Cognitive decline). تتطوّر أشكال المرض عند الشباب والبالغين بصورة أبطأ، ولكنه يؤدي في النهاية إلى ضعف عصبي كبير وإلى الوفاة[34].
[الشكل6]
 تفاعل إنزيم آريل سولفاتاز، وهو يحدث بمساعدة بروتين سابوزين ب
تفاعل إنزيم آريل سولفاتاز، وهو يحدث بمساعدة بروتين سابوزين ب
حذف الصورة؟
سيؤدي هذا إلى نقل الصورة إلى سلة المهملات.
قد يؤدي زراعة نخاع العظم، بما في ذلك زراعة
الخلايا الجذعية، إلى إبطاء تطوّر المرض
في الجهاز العصبي المركزي عند المرضى الرضّع الذين لم تظهر عليهم أعراض المرض بعد، وكذلك أولئك البالغون الذين يعانون من المرض وإن لم تظهر عليهم الأعراض، أو يعانون من أعراض خفيفة. وفي عام 2024، أعطت إدارة الغذاء والدواء الأميركية الموافقة على استخدام دواء أتيدارساجين أوتوتيمسيل (Atidarsagene autotemcel) لعلاج هذا المرض سواء قبل ظهور الأعراض أو معها. من الجدير ذكره أن الدواء نفسه موافق عليه في أوروبا منذ عام 2020. يمكن علاج حاملي الصفة المتنحية للمرض الذين لم تظهر عليهم الأعراض بعدُ بجرعة علاج واحدة من هذا الدواء[35]. هذا الدواء جزء من استراتيجية
العلاج الجيني (Gene therapy).
نمط الوراثة
تُورَّث أمراض تخزين
الدهون السفينغولية عادةً بطريقة متنحية جسمية (Autosomal recessive)، إذ يحمل كلا الوالدين أليلًا متحورًا (Mutated allele) للجين الذي يشفّر
الإنزيم الناقص. والاستثناء هو مرض فابري الذي يُورَّث بطريقة مرتبطة بالكروموسوم X. وقد حدّدت التطورات في مجال البحوث الجينية
الطفرات المسؤولة عن نقص هذه
الإنزيمات؛ ما مكّن من إجراء الاختبارات الجينية وفحص ناقلي المرض والتشخيص قبل الولادة[36].
الفيزيولوجيا المرضية والأعراض السريرية
بسبب نقص في بعض
الإنزيماتالليسوسومية، يؤدي تراكم جزيئات
الدهون السفينغولية في
الليسوسومات إلى أمراض تخزين
الدهون السفينغولية، وهي اضطرابات
أيضية موروثة. إن هذه
الإنزيمات ضرورية لتكسير
الدهون السفينغولية إلى جزيئات أصغر. تتضمن الفيزيولوجيا المرضية لأمراض التخزين هذه عدة آليات رئيسة تؤدي إلى اختلالات في
الخلايا وإلى تلف الأنسجة. يتسبب تخزين
الدهون السفينغولية المتراكمة في تعطيل الوظيفة الطبيعية للأجسام الحالّة وإعاقة تحلّل الجزيئات الكبرى الأخرى؛ ما يؤدّي إلى تخزين ثانوي لركائز إضافية. ويؤدّي التخزين المفرط للمواد إلى تضخم
الليسوسومات؛ ما يؤدي إلى اختلالات في
الخلايا وتأثيرات سلبية على معظمها؛ إذ يكبر حجمها بسبب المواد المختزنة فيها وتعاني عند تأدية وظائفها. يؤثر التراكم في
الخلايا بشكل عام، ولكن التأثير الأكبر يكون في
الخلايا العصبية بشكل خاص. عند تراكم
الدهون داخل
الخلايا، تفشل
الخلية في تأدية مهمّاتها، وفي العديد من الأحيان تموت. إن موت
الخلايا العصبية إشكالي للكائن الحي؛ فلا يمكن أن تتجدد هذه
الخلايا، وبناء عليه، إنّ جزءًا كبيرًا من الفيزيولوجيا المرضية لهذه الأمراض يتضمن موت
الخلايا العصبية، الذي يؤدي إلى أعراض مرضية عصبية جسيمة[37].
الخلل الخلوي وتلف الأنسجة العام
يؤدّي تراكم
الدهون السفينغولية في الأنسجة والأعضاء المختلفة إلى سلسلة من الأحداث المرضيّة[38]، من ضمنها:
- الالتهاب: يؤدي تراكم
الدهون السفينغولية إلى تنشيط المسارات الالتهابية؛ ما يؤدي إلى إطلاق
السيتوكينات {{السيتوكينات: فئة واسعة من البروتينات الصغيرة التي تفرزها الخلايا لتعمل بصفتها جزيئات إشارة تتوسط في التفاعلات المناعية والالتهابات وتنظمهما.}} المؤيدة للالتهابات (Pro-inflammatory cytokines) واستقطاب
الخلايا المناعية. يساهم الالتهاب المزمن، بسبب تراكم
الدهون، في تلف الأنسجة وتفاقم تطور المرض.
- الإجهاد التأكسدي: يؤدي ضعف تحلل
الدهون السفينغولية إلى توليد
مركّبات الأكسجين التفاعلية (Reactive oxygen species)؛ ما يؤدّي إلى تلف تأكسدي للمكونات الخلوية، بما في ذلك
الدهون والبروتينات والحمض النووي.
- الاستماتة الخلوية: يمكن أن يؤدّي تراكم
الدهون السفينغولية إلى تحفيز مسارات
الموت الخلوي المبرمج المختلفة؛ ما يؤدي إلى
موت الخلية {{موت الخلية: عملية بيولوجية حيوية ومستمرة تُزال فيها الخلايا التالفة أو المُصابة أو غير الضرورية، ما يُحافظ على توازن الأنسجة ونموها بشكل طبيعي. هناك العديد من المسارات المُنظّمة المؤدية للموت الخلوي، من أبرزها موت الخلايا المُبرمج، أو استماتة الخلايا (Apoptosis)، والنخر (Necrosis)، أي موت الخلايا غير المُتحكّم فيه، والالتهام الذاتي (Autophagy) أي التحلل الذاتي.}}. وهذا مهم بشكل خاص في الأشكال العصبية التنكّسية من أمراض تخزين
الدهون السفينغولية؛ إذ يؤدي موت
الخلايا العصبية إلى ظهور المظاهر السريرية العصبية.
- خلل في الحركة الخلوية الداخلية وانتقال المواد: يؤثر تعطل وظيفة
الليسوسوم في انتقال المواد داخل الخلية؛ ما يؤدي إلى سوء توطين
البروتينات والجزيئات الكبرى الأخرى، ويُضعف وظائف
الخلية المختلفة.
تلف الأنسجة والأعضاء الخاص
تتنوع الأعراض السريرية لأمراض تخزين
الدهون السفينغولية وتختلف حسب الاضطراب المحدد ونقص
الإنزيم وأنواع
الدهون السفينغولية التي تتراكم. يؤثر التراكم المرضي للدهون السفينغولية في العديد من الأعضاء والأجهزة في جسم الإنسان؛ ما يؤدي إلى أعراض سريرية مميزة[39]، تظهر في:
تشخيصها وخيارات علاجها
التشخيص والتدخل المبكران أمران أساسيان لتحسين النتائج لدى المرضى المصابين بأمراض تخزين
الدهون السفينغولية. إن الرعاية الطبية لهؤلاء المرضى عابرة للتخصصات، فهي تشمل استشارات من علماء الوراثة وأطباء الأعصاب وأطباء الدم وغيرهم من المتخصصين، لتُدار هذه الأمراض المعقدة بطريقة شاملة ومُرضية[40].
التشخيص
يتضمن تشخيص أمراض تخزين
الدهون السفينغولية في العادة مزيجًا من التقييمات السريرية والاختبارات البيوكيميائية والفحوصات الجينية. في التقييم السريري الأولي، غالبًا ما يُكشف عن علامات وأعراض مميزة لهذه الأمراض، مثل تضخم الأعضاء والعجز العصبي والمظاهر الجلدية؛ ما يستدعي إجراء المزيد من الفحوصات التأكيدية، نحو ما يأتي:
- تقنيات التصوير الطبي: يمكن لتقنيات التصوير الطبي مثل التصوير بالرنين المغناطيسي (Magnetic resonance imaging)، والتصوير المقطعي المحوسب (Computed tomography scan)، والتصوير بالموجات فوق الصوتية (Ultrasound)، الكشف عن تضخّم الأعضاء والاعتلالات العظمية والعصبية. يُعد التصوير بالرنين المغناطيسي مفيدًا بشكل خاص في تشخيص مرض كرابي وحثل المادة البيضاء المتبدل اللون، إذ يكشف عن تغيرات المادة البيضاء في
الدماغ وإزالة
الميالين في الأعصاب.
- الاختبارات البيوكيميائية: في هذه الفحوص، يقاس نشاط
إنزيم محدد في
كريات الدم البيضاء (Leukocytes) أو في
الخلايا الليفية (Fibroblasts)، فإذا كان منخفضًا يشير إلى تأكيد المرض. على سبيل المثال، يؤكد انخفاض نشاط الغلوكوسيريبروسيداز وجود مرض غوشيه، في حين يشير انخفاض نشاط
إنزيم بيتا هيكسوزأمينيداز أ إلى مرض تاي ساكس. كما يمكن فحص تراكم
الدهون السفينغولية المحددة في السوائل الجسدية باستخدام تقنيات
مطياف الكتلة (Mass spectrometry) أو
الكروماتوغرافيا (Chromatography).
- الفحوصات الجينية: في هذه الاختبارات، تُفحص
الجينات التي تشفّر
الإنزيمات المعنية بطرق الأحياء الجزيئية (Molecular biology) ويُبحث فيها عن
الطفرات. وجود
الطفرات لا يؤكد التشخيص فحسب، بل يسهّل أيضًا معرفة ناقل المرض ويسهّل التشخيص قبل الولادة. على سبيل المثال، تؤكّد
الطفرات في
جين GBA مرض غوشيه، في حين تؤكد
الطفرات في
جين GLA مرض فابري.
- الفحص النسيجي المرضي: في بعض الحالات، قد يلجأ الأطباء إلى أخذ خزعة من الأنسجة المصابة، مثل الكبد أو نخاع العظم أو الجلد، لمراقبة التغيّرات النسيجية، مثل وجود خلايا رغوية في مرض نيمان بيك أو خلايا كروية في المادة البيضاء في مرض كرابي.
خيارات العلاج
بصورة عامة، يهدف علاج هذه الأمراض الوراثية إلى إدارة الأعراض وإبطاء تقدّم المرض وتحسين نوعية الحياة[41]. وتشمل الأساليب العلاجية ما يأتي:
- العلاج الإنزيمي التعويضي: يتضمّن هذا العلاج استبدال
الإنزيم المعتلّ أو الناقص بإعطاء المريض
إنزيمات معاد تركيبها (Recombinant enzymes) عن طريق الوريد. إن هذه الاستراتيجية باهظة التكلفة، ولكنها فعّالة في مرض غوشيه ومرض فابري، إذ يقلل العلاج من تراكم
الدهون السفينغولية ويخفف الأعراض ويحسّن وظائف الأعضاء، مع أنه أقل فعالية في معالجة الأعراض العصبية، بسبب عدم قدرة
الإنزيم المُعطى على أن ينتقل عبر
الحاجز الدموي الدماغي (Blood-brain barrier).
- العلاج بتخفيض الركيزة: يقلل هذا العلاج من تصنيع
الدهون السفينغولية، ومن ثمّ يقلل من تراكمها. الخيار العلاجي أوفر من العلاج بتعويض الإنزيم، ولكنه أقل فعالية، ومع ذلك يُعدّ هذا العلاج مفيدًا بشكل خاص للمرضى الذين لا يتحمّلون العلاج التعويضي.
-
العلاج الجيني: يهدف
العلاج الجيني إلى تصحيح الخلل
الجيني الأساسي عن طريق إدخال نسخ وظيفية من
الجين المعتلّ وتصحيحه. تبشّر التطورات في تقنيات تصحيح الجينات (Gene editing technologies)، مثل كريسبر/ كاس 9 (CRISPR/ Cas9)، بعلاج هذه الأمراض وغيرها من الأمراض الوراثية بتصحيح المشكلة الرئيسة المسببة لها. تجري تجارب سريرية لعلاج هذه الأمراض، بما في ذلك حثل المادة البيضاء المتبدل اللون ومرض كرابي.
- زرع
الخلايا الجذعية المكونة للدم: تستخدم تقنية زراعة
الخلايا الجذعية المكونة للدم لتوفير مصدر
للخلايا المنتجة للإنزيمات الوظيفية. يمكن لهذه التقنية أن ترمّم الوظيفة العصبية أو تُحسّنها إذا أُجريت في وقت مبكر من مراحل المرض. تُستخدم هذه التقنية حاليًا لعلاج مرض كرابي وحثل المادة البيضاء المتبدل اللون.
- العلاجات الناشئة: البحث والتطوير مستمران لإنتاج علاجات جديدة للأمراض الوراثية، ومن ضمنها أمراض تخزين
الدهون السفينغولية. هناك مثلًا العلاج بالمرافق الدوائي، الذي يُرمّم
الإنزيمات المشوهة، والأدوية الأخرى التي تعزز وظائف
الليسوسوم.
- الرعاية الداعمة: تؤثر أمراض تخزين
الدهون السفينغولية في جودة الحياة بشكل كبير، لذلك فإن الرعاية الداعمة الشاملة ضرورية لإدارة الأعراض وتحسين نوعية الحياة. يتضمن ذلك إدارة الألم والعلاج الطبيعي والعلاج الوظيفي.
المراجع
Abed Rabbo, Muna et al. “Sphingolipid Lysosomal Storage Diseases: From Bench to Bedside.”
Lipids Health Disease. vol. 20, article no. 44 (2021).
Amaducci, Luigi et al. “The First Alzheimer Disease Case: A Metachromatic Leukodystrophy.”
Dev Neurosci. vol. 13, no. 4-5 (1991). pp. 186-187.
Austin, James H. et al. “A Controlled Study of Enzymic Activities in Three Human Disorders of Glycolipid Metabolism.”
Journal of Neurochemistry. vol. 10, no. 11 (1963). pp. 805-816.
Bajwa, Hamza & Waqas Azhar. “Niemann-Pick Disease.” in:
StatPearls. Treasure Island, FL: StatPearls Publishing, 2025. at:
https://acr.ps/1L9F2in
Brady, Roscoe O., Julian Kanfer & David Shapiro. “Metabolism of Glucocerebrosides II. Evidence of an Enzymatic Deficiency in Gaucher’s Disease.”
Biochemical & Biophysical Research Communications. vol. 18, no. 2 (1965). pp. 221-225.
Crocker, Allen. “The Cerebral Defect in Tay-Sachs Disease and Niemann-Pick Disease.” Journal of Neurochemistry. vol. 7, no. 1 (1961). pp. 69-80.
Dandana, Azza et al. “Gaucher Disease: Clinical, Biological and Therapeutic Aspects.”
Pathobiology. vol. 83, no. 1 (2016). pp. 13-23.
DeLuca, Chester, Judith A. Brown & Thomas B. Shows. “Lysosomal Arylsulfatase Deficiencies in Humans: Chromosome Assignments for Arylsulfatase A and B.”
Proceedings of the National Academy of Sciences. vol. 76, no. 4 (1979). pp. 1957-1961.
“Enzyme Replacement Therapy.”
National Gaucher Foundation. at:
https://acr.ps/1L9F2P3
Fabry, H. “An Historical Overview of Fabry Disease.”
Journal of Inherited Metabolic Disease. vol. 24, no. S2 (2001). pp. 3-7.
Fabry, Joh. “Ein Beitrag Zur Kenntniss der Purpura Haemorrhagica Nodularis (Purpura Papulosa Haemorrhagica Hebrae).”
Archiv fur Dermatologie und Syphilis. vol. 43, no. 1 (1898). pp. 187-200.
Fahim, Shahariar Mohammed et al. “Atidarsagene Autotemcel for Metachromatic Leukodystrophy.”
Journal of Managed Care & Specialty Pharmacy. vol. 30, no. 2 (2024). pp. 201-205.
Ferreira, Carlos R. & William A. Gahl. “Lysosomal Storage Diseases.”
Translational Science of Rare Diseases. vol. 2, no. 1-2 (2016). pp. 1-71.
“Gaucher Disease: Enzyme Replacement Therapy, Substrate Reduction Therapy & Potential Drug Interaction Side Effects.”
National Gaucher Foundation. at:
https://acr.ps/1L9F2pl
Gaucher, Ernest.
De l’epithélioma primitif de la rate: Hypertrophie idiopathique de la rate sans leucémie. Paris: Imprimerie Émile Martinet, 1882.
Hannun, Yusuf A. & Lina M. Obeid. “Sphingolipids and Their Metabolism in Physiology and Disease.”
Nature Reviews Molecular Cell Biology. vol. 19, no. 3 (2018). pp. 175-191.
Hannun, Yusuf A. & Lina M. Obeid. “Sphingolipids and Their Metabolism in Physiology and Disease.”
Nature Reviews Molecular Cell Biology. vol. 19, no. 3 (2018). pp. 175-191.
Jamjoum, Rama et al. “Mysterious Sphingolipids: Metabolic Interrelationships at the Center of Pathophysiology.”
Frontiers in Physiology. vol. 14, article no. 1229108 (2024).
Jatzkewitz, Horst. “Zwei Typen von Cerebrosid-Schwefelsäureestern Als Sog. Prälipoide Und Speichersubstanzen Bei Der Leukodystrophie, Typ Scholz (Metachromatische Form Der Diffusen Sklerose).”
Biological Chemistry. vol. 311, no. 4-6 (1958). pp. 279-282.
Kaguni, Laurie & Fuyuhiko Tamanoi (eds.).
History of The Enzymes, Current Topics and Future Perspectives. Cambridge: Academic press, 2023.
Kint, J. “Fabry’s Disease: Alpha-Galactosidase Deficiency.”
Science. vol. 167, no. 3922 (1970). pp. 1268-1269.
“Krabbe Disease.”
MedlinePlus. at:
https://acr.ps/1L9F2Mj
Lenders, Malte & Eva Brand. “Fabry Disease: The Current Treatment Landscape.”
Drugs. vol. 81, no. 6 (2021). pp. 635-645.
Lipiński, Patryk & Anna Tylki-Szymańska. “The Liver and Lysosomal Storage Diseases: From Pathophysiology to Clinical Presentation, Diagnostics, and Treatment.”
Diagnostics. vol. 14, no. 12, articles no. 1299 (2024).
McCafferty, Emma H. & Lesley J. Scott. “Migalastat: A Review in Fabry Disease.”
Drugs. vol. 79, no. 1 (2019). pp. 543-554.
Mistry, Pramod k. et al. “Gaucher Disease: Progress and Ongoing Challenges.”
Molecular Genetics & Metabolism. vol. 120, no. 1-2 (2017). pp. 8-21.
Okada, Shintaro & John O’Brien. “Tay-Sachs Disease: Generalized Absence of a Beta-D-N-Acetylhexosaminidase Component.”
Science. vol. 165, no. 3894 (1969). pp. 698-700.
Peiffer, Jurgen. “Über Die Metachromatischen Leukodystrophien (Typ Scholz).”
Archiv Für Psychiatrie Und Nervenkrankheiten. vol. 199, no. 4 (1959). pp. 386-416.
Pick, Ludwig. “Der Morbus Gaucher Und Die Ihm Ähnlichen Krankheiten (Die Lipoidzellige Splenohepatomegalie Typus Niemann Und Die Diabetische Lipoidzellenhypoplasie Der Milz).”
Ergebenisse Der Inneren Medizin Und Kingderheilkunde. vol. 29 (1926). pp. 519-527.
Platt, Frances et al. “Lysosomal Storage Diseases.” Nature Reviews Disease Primers. vol. 4, article no. 27 (2018).
Sachs, Bernard. “On Arrested Cerebral Development, with Special Reference to Its Cortical Pathology.”
The Journal of Nervous & Mental Disease. vol. 14, no. 9 (1887). pp. 541-553. at:
https://acr.ps/1L9F2bD
Scholz, Willibald. “Klinische, Pathologisch-Anatomische Und Erbbiologische Untersuchungen Bei Familiärer, Diffuser Hirnsklerose Im Kindesalter.” Zeitschrift Für Die Gesamte Neurologie Und Psychiatrie. vol. 99, no. 1 (1925). pp. 651-717.
Siegel, George J. et al. (eds.).
Basic Neurochemistry: Molecular, Cellular and Medical Aspects. 6th ed. Philadelphia, PA: Lippincott-Raven, 1999. at:
https://acr.ps/1L9F2hI
Sun, Angela. “Lysosomal Storage Disease Overview.”
Annals of Translational Medicine. vol. 6, no. 24 (2018).
Suzuki, Yoshiyuki & Kunihiko Suzuki. “Krabbe’s Globoid Cell Leukodystrophy: Deficiency of Galactocerebrosidase in Serum, Leukocytes, and Fibroblasts.”
Science. vol. 171, no. 3966 (1971). pp. 73-75.
Tay, Waren. “Symmetrical Changes in the Region of the Yellow Spot in Each Eye of an Infant.”
Jama Neurology. vol. 20, no. 1 (1969). pp. 104-106.
[1] Muna Abed Rabbo et al., “Sphingolipid Lysosomal Storage Diseases: From Bench to Bedside,”
Lipids Health Disease, vol. 20, article no. 44 (2021).
[2] Angela Sun, “Lysosomal Storage Disease Overview,”
Annals of Translational Medicine, vol. 6, no. 24 (2018).
[3] Rama Jamjoum et al., “Mysterious Sphingolipids: Metabolic Interrelationships at the Center of Pathophysiology,”
Frontiers in Physiology, vol. 14, article no. 1229108 (2024).; Batoul Issleny et al., “Chapter Seven - Sphingolipids: From Structural Components to Signaling Hubs,” in: Laurie Kaguni & Fuyuhiko Tamanoi (eds.),
History of The Enzymes, Current Topics and Future Perspectives, vol. 54 (Cambridge: Academic press, 2023), pp. 171-201.
[4] Ernest Gaucher,
De l’epithélioma primitif de la rate: Hypertrophie idiopathique de la rate sans leucémie (Paris: Imprimerie Émile Martinet, 1882), p. 31.
[5] Roscoe O. Brady, Julian Kanfer & David Shapiro, “Metabolism of Glucocerebrosides II. Evidence of an Enzymatic Deficiency in Gaucher’s Disease,”
Biochemical & Biophysical Research Communications, vol. 18, no. 2 (1965), pp. 221-225.
[6] Waren Tay, “Symmetrical Changes in the Region of the Yellow Spot in Each Eye of an Infant,”
Jama Neurology, vol. 20, no. 1 (1969), pp. 104-106.
[7] Bernard Sachs, “On Arrested Cerebral Development, with Special Reference to Its Cortical Pathology,”
The Journal of Nervous & Mental Disease, vol. 14, no. 9 (1887), pp. 541-553, at:
https://acr.ps/1L9F2bD
[8] Shintaro Okada & John O’Brien, “Tay-Sachs Disease: Generalized Absence of a Beta-D-N-Acetylhexosaminidase Component,”
Science, vol. 165, no. 3894 (1969), pp. 698-700.
[9] Joh Fabry, “Ein Beitrag Zur Kenntniss der Purpura Haemorrhagica Nodularis (Purpura Papulosa Haemorrhagica Hebrae),”
Archiv fur Dermatologie und Syphilis, vol. 43, no. 1 (1898), pp. 187-200.
[10] J. Kint, “Fabry’s Disease: Alpha-Galactosidase Deficiency,”
Science, vol. 167, no. 3922 (1970), pp. 1268-1269.
[11] Luigi Amaducci et al., “The First Alzheimer Disease Case: A Metachromatic Leukodystrophy,”
Dev Neurosci, vol. 13, no. 4-5 (1991), pp. 186-187.
[12] Willibald Scholz, “Klinische, Pathologisch-Anatomische Und Erbbiologische Untersuchungen Bei Familiärer, Diffuser Hirnsklerose Im Kindesalter,” Zeitschrift Für Die Gesamte Neurologie Und Psychiatrie, vol. 99, no. 1 (1925), pp. 651-717.
[13] Jurgen Peiffer, “Über Die Metachromatischen Leukodystrophien (Typ Scholz),”
Archiv Für Psychiatrie Und Nervenkrankheiten, vol. 199, no. 4 (1959), pp. 386-416.
[14] Horst Jatzkewitz, “Zwei Typen von Cerebrosid-Schwefelsäureestern Als Sog. Prälipoide Und Speichersubstanzen Bei Der Leukodystrophie, Typ Scholz (Metachromatische Form Der Diffusen Sklerose),”
Biological Chemistry, vol. 311, no. 4-6 (1958), pp. 279-282.
[15] James H. Austin et al., “A Controlled Study of Enzymic Activities in Three Human Disorders of Glycolipid Metabolism,”
Journal of Neurochemistry, vol. 10, no. 11 (1963), pp. 805-816.
[16] Ludwig Pick, “Der Morbus Gaucher Und Die Ihm Ähnlichen Krankheiten (Die Lipoidzellige Splenohepatomegalie Typus Niemann Und Die Diabetische Lipoidzellenhypoplasie Der Milz),”
Ergebenisse Der Inneren Medizin Und Kingderheilkunde, vol. 29 (1926). pp. 519-527.
[17] Ibid.
[18] Allen Crocker, “The Cerebral Defect in Tay-Sachs Disease and Niemann-Pick Disease,” Journal of Neurochemistry, vol. 7, no. 1 (1961), pp. 69-80.
[19] “Krabbe Disease,”
MedlinePlus, accessed on 21/2/2025, at:
https://acr.ps/1L9F2Mj
[20] Yoshiyuki Suzuki & Kunihiko Suzuki, “Krabbe’s Globoid Cell Leukodystrophy: Deficiency of Galactocerebrosidase in Serum, Leukocytes, and Fibroblasts,”
Science, vol. 171, no. 3966 (1971), pp. 73-75.
[21] Frances Platt et al., “Lysosomal Storage Diseases,” Nature Reviews Disease Primers, vol. 4, article no. 27 (2018).
[22] Platt et al.
[23]في بعض الأحيان يعتلّ أكثر من إنزيم ليؤدي إلى المرض نفسه، كما يمكن أيضًا للمرض أن يظهر بسبب اختلال في بعض البروتينات المساندة مثل السابوزينات (Saposins)
[24]تتراكم مواد وجزيئات أخرى، ولكن هذه المركّبات هي السائدة.
[25] Azza Dandana et al., “Gaucher Disease: Clinical, Biological and Therapeutic Aspects,”
Pathobiology, vol. 83, no. 1 (2016), pp. 13-23.
[26] Pramod k. Mistry et al., “Gaucher Disease: Progress and Ongoing Challenges,”
Molecular Genetics & Metabolism, vol. 120, no. 1-2 (2017), pp. 8-21.
[27] “Enzyme Replacement Therapy,”
National Gaucher Foundation, accessed on 8/6/2025, at:
https://acr.ps/1L9F2P3
[28] “Gaucher Disease: Enzyme Replacement Therapy, Substrate Reduction Therapy & Potential Drug Interaction Side Effects,”
National Gaucher Foundation, accessed on 8/6/2025, at:
https://acr.ps/1L9F2pl
[29] H. Fabry, “An Historical Overview of Fabry Disease,”
Journal of Inherited Metabolic Disease, vol. 24, no. S2 (2001), pp. 3-7.
[30] Malte Lenders & Eva Brand, “Fabry Disease: The Current Treatment Landscape,”
Drugs, vol. 81, no. 6 (2021), pp. 635-645.
[31] Emma H. McCafferty & Lesley J. Scott, “Migalastat: A Review in Fabry Disease,”
Drugs, vol. 79, no. 1 (2019), pp. 543-554.
[32] Hamza Bajwa & Waqas Azhar, “Niemann-Pick Disease,” in:
StatPearls (Treasure Island, FL: StatPearls Publishing, 2025), accessed on 24/8/2025, at:
https://acr.ps/1L9F2in
[33] “Krabbe Disease,”
[34] Chester DeLuca, Judith A. Brown & Thomas B. Shows, “Lysosomal Arylsulfatase Deficiencies in Humans: Chromosome Assignments for Arylsulfatase A and B,”
Proceedings of the National Academy of Sciences, vol. 76, no. 4 (1979), pp. 1957-1961.
[35] Shahariar Mohammed Fahim et al., “Atidarsagene Autotemcel for Metachromatic Leukodystrophy,”
Journal of Managed Care & Specialty Pharmacy, vol. 30, no. 2 (2024), pp. 201-205.
[36] Kunihiko Suzuki & Marie T. Vanier, “Lysosomal Disease,” in: George J. Siegel et al. (eds.),
Basic Neurochemistry: Molecular, Cellular and Medical Aspects, 6th ed. (Philadelphia, PA: Lippincott-Raven, 1999), at:
https://acr.ps/1L9F2hI
[37] Patryk Lipiński & Anna Tylki-Szymańska, “The Liver and Lysosomal Storage Diseases: From Pathophysiology to Clinical Presentation, Diagnostics, and Treatment,”
Diagnostics, vol. 14, no. 12, articles no. 1299 (2024).
[38] Yusuf A. Hannun & Lina M. Obeid, “Sphingolipids and Their Metabolism in Physiology and Disease,”
Nature Reviews Molecular Cell Biology, vol. 19, no. 3 (2018), pp. 175-191.
[39] Platt et al.
[40] Carlos R. Ferreira & William A. Gahl, “Lysosomal Storage Diseases,”
Translational Science of Rare Diseases, vol. 2, no. 1-2 (2016), pp. 1-71.
[41] Lipiński & Tylki-Szymańska.
