الانبثاث أو النقائل هو نمو الخلايا السرطانية في أعضاء بعيدة عن العضو الذي نشأت فيه، وهي آخر مرحلة تصل إليها الخلايا السرطانية، لذلك تُعدّ الصفة الأكثر فتكًا. تحدث الوفاة لدى الغالبية العظمى من مرضى السرطان بسبب وصول مرضهم إلى مرحلة الانبثاث، وليس بسبب الأورام الأساسية {{الورم الأساسي (Primary tumors): الورم الذي ينشأ في العضو أو النسيج نفسه، وليس ناتجاً عن انبثاث (Metastasis) من عضو آخر.}}. يشمل الانبثاث سلسلة من الأحداث البيولوجية إذ تكتسب خلايا الورم الأساسي تدريجيًّا القدرة على الغزو (Invasion) عبر الغشاء القاعدي {{الغشاء القاعدي (Basement membrane): طبقة رقيقة من المادة خارج الخلوية (Extracellular matrix)، تفصل بين الخلايا الطلائية والنسيج الضام.}} إلى الأنسجة الأعمق؛ ثم تنتشر عبر الدم أو الجهاز اللمفاوي (Lymphatic system) أو عن طريق الاختراق المباشر للأنسجة المجاورة لتستقر في الأعضاء البعيدة، وتستأنف في النهاية التكاثر في مواقع بعيدة لاستعمار هذه الأعضاء.
تدفع كلٌّ من هذه الأحداث بقدرة الخلايا السرطانية على تبني حالات خلوية مختلفة، واستقطاب الخلايا المناعية (Immune cells) واللحمية {{اللحمية (Stromal cells): هي الخلايا الداعمة الموجودة في السدى، وهو النسيج الضام الذي يحيط بالخلايا المتخصصة في أي عضو.}} المحيطة بها في بيئة الورم لدعم نموها والتهرُّب من الجهاز المناعي (Immune system). وعلى عكس الأورام الأولية التي يمكن في الغالب علاجها باستخدام علاجات موضعية مثل الجراحة والإشعاع، فإن سرطان النقائل (Metastatic cancer) هو مرض جهازي يصيب أعضاء متعددة، إما عن طريق استعمار الأعضاء بشكل مباشر وإعاقة وظيفتها، أو عن طريق تغيير تفاعلات الاستقلاب {{تفاعلات الاستقلاب (Metabolic reactions): مجموعة التفاعلات الكيميائية الحيوية التي تحدث داخل الخلايا للحفاظ على الحياة، وتساعد في إنتاج الطاقة اللازمة للعمليات الحيوية، كما وتُنتج من خلالها مركبات جديدة تُستعمل في بناء الخلايا والأنسجة.}} فيها من خلال إفرازات معدَّلة، ما يؤدي في النهاية إلى موت المريض. عندما يصل السرطان إلى مرحلة النقائل، فإن هذه الظاهرة غير قابلة للشفاء سريريًّا إلى حد كبير مع استثناءات قليلة، وذلك بسبب اكتساب الأورام النقيلية خصائص مقاومة للعلاجات المتاحة حتى نهاية 2024.
مراحله
يمكن تقسيم الانبثاث إلى ثلاث مراحل من الممكن أن تتداخل زمنيًّا: الانتشار (Dissemination)، الكُمُون (Dormancy)، والاستعمار (Colonization)، إذ تخضع الخلايا السرطانية لسلسلة من الخطوات لغزو الأنسجة والبقاء على قيد الحياة أثناء الانتقال واستعمار الأعضاء، ويطلق على هذه المراحل مجتمعةً سلسلة تتالي الانبثاث (Metastasis cascade)[1] (الشكل 1). من العوامل الأولى التي تسهم في بدء هذه العملية التغييرات التي تحصل في المصفوفة خارج الخلية {{المصفوفة خارج الخلية (Extracellular matrix): شبكة من الجزيئات الكبيرة موجودة خارج الخلايا، تُشكّل الوسط الذي تعيش فيه الخلايا، وتتحرك وتتواصل فيه، وتعمل على دعمها.}}[2] حول الخلايا السرطانية.
[الشكل 1]
 سلسلة أحداث تتالي الانبثاث
سلسلة أحداث تتالي الانبثاث
حذف الصورة؟
سيؤدي هذا إلى نقل الصورة إلى سلة المهملات.
خلال مرحلة الانتشار، تغزو الخلايا السرطانية، وهي تحمل طفرات مسرطنة، الغشاء القاعدي إلى طبقات أعمق من الأنسجة، مكتسبة القدرة على البقاء على قيد الحياة والنمو بدون الحاجة إلى عوامل النمو {{عوامل النمو (Growth factors): بروتينات طبيعية تُنتجها الخلايا، وتعمل كإشارات كيميائية تُنظّم نمو الخلايا، و انقسامها، وتكاثرها، وتمايزها (تحوّلها إلى نوع متخصص)، وبقائها أو موتها المبرمج.}}، التي يحتاجها النسيج الأصلي للنمو. بعدها تخترق هذه الخلايا الأوعية الدموية أو اللمفاوية القريبة في عمليّة تُسمى دخول الوعاء (Intravasation) من خلال الهجرة عبر الخلايا البطانية (بطانة الأوعية الدموية) واختلال الشعيرات الدموية، أو الهجرة على طول الأعصاب، أو الانتشار المباشر المحلي إلى الفراغات المجاورة، مثل تجويف البطن (Abdominal cavity) أو الجنب (Pleural cavity)، وبعد دخولها في الأوعية تمشي في مجرى الدم أو السائل اللمفاوي وتخرج إلى الأعضاء البعيدة[3].
في الدورة الدموية، تعاني الخلايا السرطانية المنتشرة من ظروف غير مثالية بسبب العوامل الفيزيائية والاختزالية والمناعية الموجودة[4]. تنتشر الخلايا السرطانية على صورة خلايا فردية أو في مجموعات صغيرة غنية بخلايا سرطانية تشبه الخلايا الجذعية (Stem cells)، مغطّاة بالصفائح الدموية {{الصفائح الدموية: أجزاء صغيرة من خلايا دموية صغيرة جدًا، تنشأ من انقسام خلايا موجودة في نخاع العظم تُسمى الخلايا العملاقة، وتكمن وظيفتها في المساهمة في تخثر الدم لمنع النزيف من الأوعية الدموية المقطوعة.}} (Platelets) أو الخلايا العَدِلة (Neutrophils) أو الخلايا اللُحمية المشتقة من الورم (Stromal tumor cells)، ما قد يحميها من المراقبة المناعية وتمنح مجموعات الخلايا السرطانية المنتشرة في الدم قدرة أكبر على الانبثاث مقارنة بالخلايا الفردية المعرضّة أكثر للتدمير بوساطة الجهاز المناعي[5] (الشكل 2).
بمجرد أن تصل إلى الأعضاء البعيدة، يُقضى على الخلايا السرطانية المنتشرة بشكل أكبر بسبب الإجهاد التأكسدي (Oxidative stress) العالي، ونقص عوامل النمو أو المغذيات الداعمة، وكثرة الدفاعات المناعية المتمثلة في الخلايا البلعمية (Macrophages) الخاصة بالأنسجة والخلايا الفاتكة الطبيعية {{الخلايا الفاتكة الطبيعية (Natural killer cells): نوع من خلايا الجهاز المناعي الفطريّ، تؤدي دورًا مهمًا في مهاجمة الخلايا المصابة بالفيروسات أو الخلايا السرطانية، من دون الحاجة إلى تعرّف مسبق على المستضد، فهي تتعرف على الخلايا غير الطبيعية ثم تدمرها.}} والخلايا اللمفاوية التائية {{الخلايا اللمفاوية التائية (T cell lymphocytes): نوع من خلايا الدم البيضاء، تؤدي دورًا رئيسًا في الجهاز المناعي التكيفي، تُنتج في نخاع العظم، ثم تنتقل إلى الغدة الزعترية لتنضج وتصبح قادرة على التمييز بين الخلايا السليمة والخلايا الغريبة.}} وغيرها من آليات المراقبة المناعية[6]. يمكن للخلايا السرطانية المنتشرة الباقية الدخول في فترة كمون متغيرة المدة، فإما أن تخرج من دورة الخلية {{دورة الخلية (Cell cycle): سلسلة من الأحداث التي تمر بها الخلية للنمو، والانقسام، وإنتاج خلايا جديدة. تنقسم دورة الخلية إلى مراحل منظمة لضمان النسخ الصحيح للحمض النووي (DNA)، والانقسام المتساوي للخلايا.}} وتكمن ساكنةً، أو أن تدخل في حالة توازن ديناميكي فيه نوبات سريعة من التكاثر يقابلها حركة مناعية قوية تؤدي إلى التخلص من هذه الخلايا، أو احتواء الخلايا الورمية المتكاثرة من قبل البيئة المحيطة بالورم، بحيث يكون هناك بالمحصلة القليل من النمو الانبثاثي[7]. يُعدّ الانتشار والكمون انبثاثًا فرعيًّا لأن الخلايا السرطانية المنتشرة غير قابلة للكشف بالتصوير السريري ولا يدرك المرضى الإصابة بالمرض الفرعي (سرطان غير مشخص سريريًّا).
[الشكل 2]
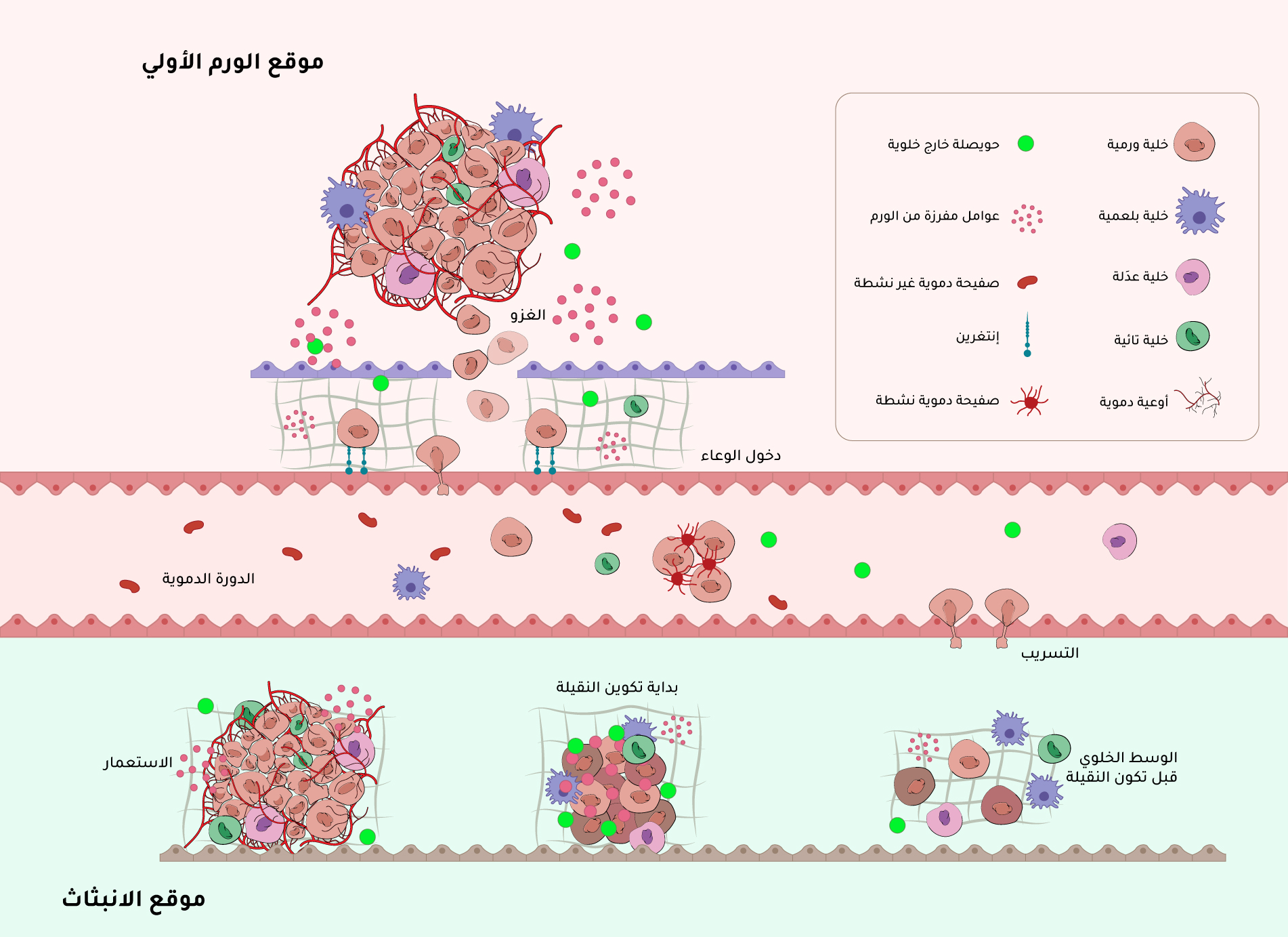 مراحل الانبثاث وغزو الخلايا السرطانية
مراحل الانبثاث وغزو الخلايا السرطانية
حذف الصورة؟
سيؤدي هذا إلى نقل الصورة إلى سلة المهملات.
تُستمد النقائل الكبيرة الظاهرة سريريًّا من خلايا الانبثاث الناجحة التي تكيّفت واستقطبت بيئتها المحيطة بالورم لتتيح في النهاية النمو واستعمار الأعضاء عن طريق تحفيز مسارات تجديد الأنسجة، وتكوين الأوعية الدموية {{تكوين الأوعية الدموية (Angiogenesis): عملية تكوّن أو نمو أوعية دموية جديدة من أوعية دموية موجودة مسبقًا، وتعدّ عملية أساسية لنمو الأنسجة وتجديدها، وتزويدها بالأكسجين والمواد الغذائية، والتخلص من الفضلات.}}، وتثبيط المناعة. تمثل سلسلة تتابع أحداث الانبثاث استمرارية لإعادة البرمجة الخلوية والبيئية المحيطة بالورم والانتقاء الاستنساخي {{الانتقاء الاستنساخي (Clonal selection): نظرية أساسية في علم المناعة، تشرح كيفية استجابة الجهاز المناعي التكيفي لوجود مسببات الأمراض، وكيفية تطوّر خلايا الذاكرة المناعية وتكاثرها للاستجابة لها. وقد طرح هذه النظرية لأول مرة العالم الأسترالي فرانك ماكفارلين بورنت عام 1957.}} للسلالات الفرعية من الخلايا السرطانية القادرة على تحمل ضغوط البيئة المحيطة الانتقائية[8]. يؤدي هذا إلى نمو ورمي غير مقيّد، ما يؤدي إلى اختلال وظائف الأعضاء وانهيار وظيفة أجهزة الكائن الحي، وفي النهاية موت المريض.
انتشار الخلايا السرطانية وغزوها
يسبق انتشار الخلايا السرطانية خطوات أولية تحضّر للغزو والانبثاث[9]. من الأسباب الرئيسة عدم الاستقرار الكروموسومي {{عدم الاستقرار الكروموسومي (Chromosomal instability): حالة تتميّز بارتفاع معدل الطفرات في المادة الوراثية، مثل الحذف، أو التكرار، أو الانتقال من مكان إلى آخر على طول سلسلة الحمض النووي، ما يؤدي غالبًا إلى تطوّر السرطان.}} الناجم عن أخطاء متتالية في تضاعف الكروموسومات وانفصالها أثناء عمليات الانقسام المتساوي {{الانقسام المتساوي (الميتوزي) (Mitosis): عملية انقسام تحدث في الخلايا حقيقية النواة، تؤدي إلى إنتاج خليتين متماثلتين وراثيًّا، تمثل كل منهما نسخة عن الخلية الأصلية. خلال هذه العملية، تتضاعف كروموسومات الخلية الأصلية، ثم تنقسم بالتساوي بين الخليتين الناتجتين.}}. تتسبب الأخطاء في انفصال الكروموسومات في إنتاج أنوية صغيرة (Micronuclei) وتمزّقها وإفراز الحمض النووي منها إلى السيتوبلازم (Cytoplasm) (الشكل 3)، ما يؤدي بعد ذلك إلى تنشيط مسار الاستشعار بالحمض النووي في السيتوبلازم (Cytosolic DNA sensing pathway)، وهو مسار أساسي في جهاز المناعة الفطري يكشف عن الحمض النووي في السيتوسول لتحفيز إنتاج الإنترفيرونات من النوع الأول لتعزيز الاستجابة المناعية.
في السرطان، يمكن تنشيط هذا مسار بواسطة الحمض النووي المفرز داخل الخلايا السرطانية، ما يؤدي إلى تحفيز الاستجابات المناعية التي قد تسهم في قمع نمو الورم التي تؤدي إلى إطلاق إشارات العامل النووي المعزز لسلسلة كابا الخفيفة في الخلايا البائية النشطة (Nuclear factor kappa-light-chain-enhancer of activated B cells, NF-κB)[10]. يُستخدم NF-κB في خلايا حقيقية النوى منِّظمًا للجينات التي تتحكم في تكاثر الخلايا وبقائها على قيد الحياة. وبناءً عليه، فإن العديد من أنواع الأورام تعاني من اختلال في تنظيمه. يُفعّل النشاط الزائد لهذا البروتين التعبير عن جينات تحافظ على تكاثر الخلايا وتحميها من الظروف التي قد تؤدي في العادة إلى موتها بوساطة مسار الاستماتة الخلوية (Apoptosis).
[الشكل 3]
 عدم الاستقرار الكروموسومي وتكوين الأنوية الصغيرة
عدم الاستقرار الكروموسومي وتكوين الأنوية الصغيرة
حذف الصورة؟
سيؤدي هذا إلى نقل الصورة إلى سلة المهملات.
تشير الدراسات إلى أن طبيعة الخلية السرطانية البذرية الأساسية تحدد الخصائص الانبثاثية المختلفة فيما يتعلق بالنمو والاستجابة للعلاج[11]. تُظهر الدراسات الحية داخل جسم الفئران (in vivo) أو خارجه (في المختبرin vitro ) أن الخلايا السرطانية الانبثاثية تهاجر بشكل فردي[12]. ومع ذلك، يُعتقد أن البذر (Seeding) في البشر يتطلب تحرك مجموعة من الخلايا السرطانية معًا[13]، وهو ما يجعل دراسة عملية التحول من الخلايا الطلائية (الظهارية Epithelial cells) إلى خلايا اللُحمة المتوسطية (الميزينكيماMesenchymal cells ) ضرورية.
التحول الطلائي اللُحمي المتوسط
الخلايا الطلائية خلايا غير متحركة ومرتبطة بإحكام ببعضها وبالمصفوفة خارج الخلية[14]، في حين أن خصائص خلايا اللحمة المتوسطية تختلف؛ فهي خلايا متحرّكة ولديها القدرة على الانتقال والغزو. في بعض الحالات أثناء تطور الجنين، تستطيع بعض الخلايا الطلائية أن تتحول إلى خلايا اللحمة المتوسطية من خلال عملية التحول الطلائي اللُحمي المتوسط (Epithelial-mesenchymal transition)، وهي عملية بيولوجية تُفقد الخلايا الطلائية (الظهارية) خواص الالتصاق بين الخلايا والثبات في مكانها، وتكتسب خصائص خلايا اللحمة المتوسطية، مثل زيادة الحركة والقدرة على الغزو والهجرة. وفي هذه العملية، تكتسب الخلايا الطلائية المتحولة القدرة على مقاومة الإجهاد والغزو والانتشار من مكانها إلى أماكن مجاورة، وقد تصل إلى أماكن بعيدة إذا وصلت إلى مجرى الدم[15] (الشكل 3). هذه العملية ضرورية لتطور السرطان. ومع أن جزءًا من خلايا الورم الأساسي الطلائية تتحول في هذه العملية، إلا أن معظم الخلايا في الورم الرئيس لا تسهم في تطور النقائل.
[الشكل 4]
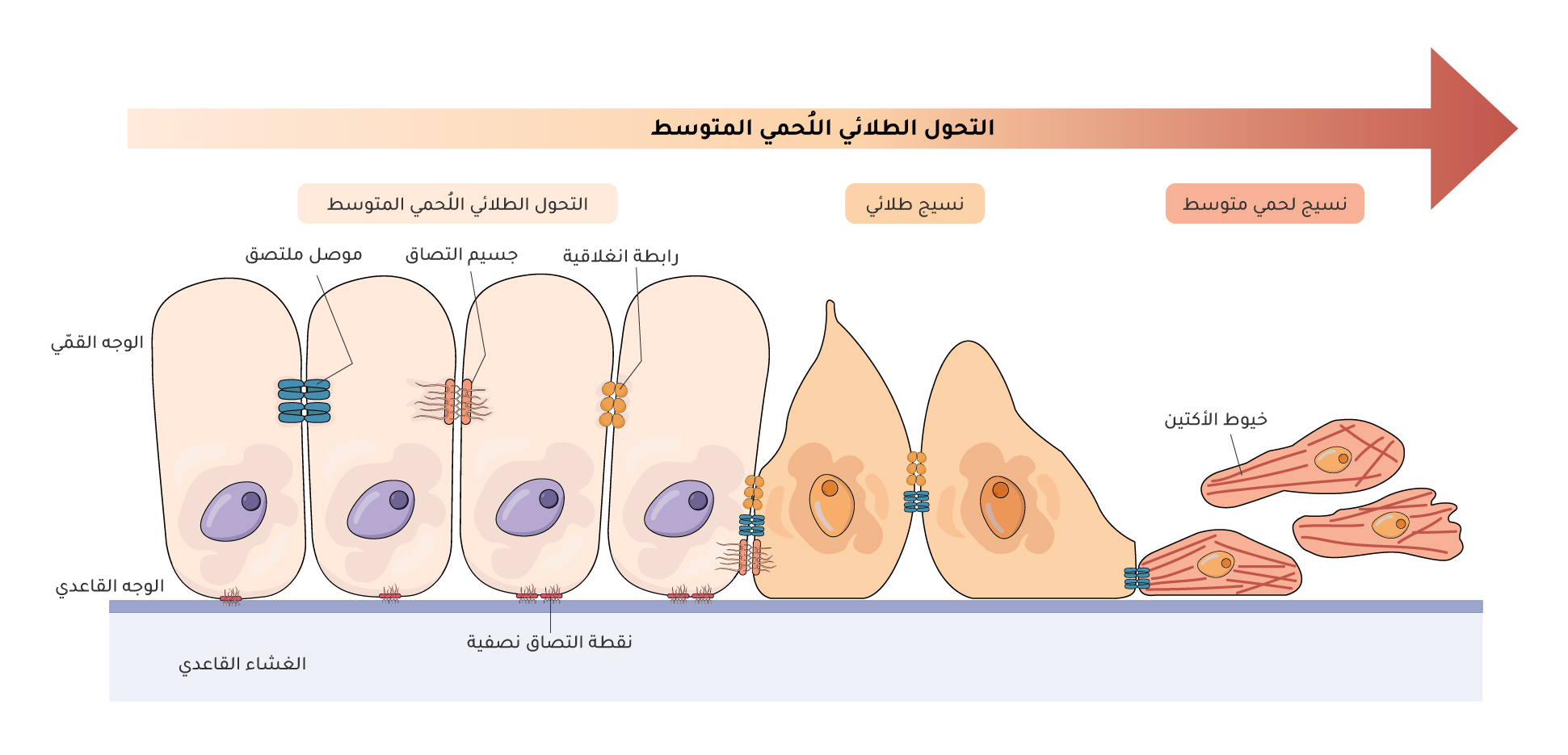 التحول الطلائي اللُحمي المتوسط
التحول الطلائي اللُحمي المتوسط
حذف الصورة؟
سيؤدي هذا إلى نقل الصورة إلى سلة المهملات.
يحكم عملية التحول الطلائي اللُحمي المتوسط تغيرات بيوكيميائية قابلة للانعكاس، تسمح للخلية الطلائية بالحصول على نمط خلية اللحمة المتوسطية[16]. وفي دراسة أجريت عام 2018 ونشرت في مجلة الطبيعة (Nature) أن الحمض الأميني الأسباراجين (Asparagine) يحفّز تكوين النقائل من خلال تحفيز الإنزيم المصنّع له الأسباراجين سينثاز (Asparagine synthase)، أما تثبيط مستويات هذا الحمض الأميني من خلال تحفيز الإنزيم الهادم له، الأسباراجيناز (Asparaginase) أو من خلال تقييد النظام الغذائي يؤدي إلى تقليل الانتشار النقيلي[17]. بالإضافة إلى ذلك، يحفّز نقص الأكسجين[18] والإجهاد الأيضي في الخلايا السرطانية على التحول الطلائي اللُحمي المتوسط[19]. وغالبًا ما يقود هذا التحول عوامل النسخ (Transcription factors) المبرمجة لتثبيط جينات الخلايا الطلائية وتفعيل جينات اللحمة المتوسطة[20]. وتؤدي منظِّمات ما بعد الترجمة {{منظِّمات ما بعد الترجمة (Posttranslational regulation): دراسة التغيرات في التعبير الجيني ووظيفة الخلايا، والتي لا تشمل تغييرات في تسلسل الحمض النووي، بل تتأثر في العوامل البيئية والآليات الخلوية المختلفة.}}، والعوامل فوق الجينية {{العوامل فوق الجينية (Epigenetics): تُعدّ التغيرات الجينية المصاحبة للتقدّم في العمر من أبرز العوامل فوق الجينية. كما أن التغيرات الفيزيائية المرتبطة بالعمر في المصفوفة خارج الخلية يمكن أن تعزز أو تمنع حركة الخلايا السرطانية وقدرتها على الغزو والانبثاث.}}[21] أيضًا دورًا حيويًّا في التحكم في عملية التحول الطلائي اللُحمي المتوسط[22].
إن التحول الطلائي اللحمي المتوسط طيف من المراحل الانتقالية بين شكل الخلية الطلائيةواللحمة المتوسطية[23] (الشكل 4). تنظّم عدد من عوامل النمو[24] ومسارات الإشارة (Signaling pathways) الانتقال من حالة إلى أخرى في هذا الطيف من الطرز الشكلية {{الطراز الشكلي (Phenotypes): السمات الفيزيائية والبيوكيميائية والسلوكية الظاهرة على الكائن الحي، والتي تنشأ من تركيبته الجينية (الطراز الجيني)، وتفاعلها مع العوامل البيئية.}}[25]. إن التحول الطلائي اللحمي المتوسط الجزئي في الخلايا السرطانية الأولية ينتقل بين مراحل وسيطة لها خصائص غزويّة وانبثاثية وتمايزية مختلفة[26]. إن الخلايا السرطانية التي تمتلك مزيجًا من الطرازين الشكليين الطلائي واللحمة المتوسطية أكثر فاعلية في الدورة الدموية والاستعمار في الموقع الثانوي وتطور النقائل[27].
تمتلك المراحل المختلفة خصائص خلوية متنوعة وتعابير جينية، مختلفة تنظِّمها عوامل نسخ ومسارات إشارة مشتركة ومتميزة. تتكاثر الخلايا النقيلية ذات الطراز الشكلي اللحمة المتوسطية بالقرب من الخلايا البطانية والالتهابية. تُطلِق هذه الخلايا السرطانية كميات كبيرة من الكيموكينات {{الكيموكينات (Chemokines): مجموعة من البروتينات الصغيرة التي توجّه حركة الخلايا المناعية نحو مواقع العدوى أو الالتهاب أو الإصابة، من خلال الارتباط بمستقبِلات محددة على الخلايا المستهدفة.}} والبروتينات لجذب الخلايا المناعية وتحفيز تكوين الأوعية الدموية، ما يشكّل موضعًا التهابيًّا ممتلئًا بالأوعية الدموية لتغذية الخلايا وخدمتها[28]. كما ثبت أن الخلايا الليفية (Fibroblasts) المرتبطة بالسرطان تدفع وتوجه حركة الخلايا السرطانية من خلال بروتين فيبرونيكتين (Fibronectin)[29].
[الشكل 5]
 تنوّع حالات التحول الطلائي اللحمي المتوسط. هناك طيف من الطرز الشكليةللخلايا الطلائية واللحمة المتوسطية
تنوّع حالات التحول الطلائي اللحمي المتوسط. هناك طيف من الطرز الشكليةللخلايا الطلائية واللحمة المتوسطية
حذف الصورة؟
سيؤدي هذا إلى نقل الصورة إلى سلة المهملات.
اختراق الخلايا السرطانية للأوعية الدموية
إن دخول الوعاء هو العملية التي تخترق من خلالها الخلايا السرطانية من الورم الأساسي إلى مجرى الدم أو الجهاز اللمفاوي، ما يتيح انتشار النقائل (الشكل 5). فتنتشر الخلايا السرطانية إلى الأعضاء عبر تجويف الأوعية الدموية أو اللمفاوية، وقد يحدث بطريقة نشطة أو سلبية[30]، ويعتمد ذلك على نوع الورم والبيئة المحيطة والأوعية الدموية[31]. يُظهر نموذج الموائع الدقيقة ثلاثي الأبعاد (3D Microfluidic model) أن البطانة تُشكّل حاجزًا أمام اختراق الخلايا السرطانية للأوعية الدموية، وتُنظَّم هذه العملية بوساطة عوامل موجودة في البيئة المحيطة بالورم[32]. علاوة على ذلك، تفرض القيود البنائية للأنسجة بعض الضغوط الميكانيكية على الخلايا السرطانية الغازية في أثناء هذه العملية[33]، ما يؤدي إلى انضغاط النواة، ما يسبب تحديًا خاصًا على سلامة نواة الخلية الغازية، وهذا يتسبب في حدوث إعادة ترتيب جيني، ما يزيد من الإمكانات الانبثاثية[34].
 [الشكل 6]
[الشكل 6]
حذف الصورة؟
سيؤدي هذا إلى نقل الصورة إلى سلة المهملات.
تسهم بروتينات الإنتغرين (Integrins)، وهي مستقبلات التصاق خلوي رئيسة في الخلايا، في كل خطوة تقريبًا من تطور السرطان بدءًا من تطور الورم الأساسي إلى حدوث الانبثاث[35]. تُظهر دراسات عديدة التغييرات في التعبير الجيني للإنتغرين في الأورام، إذ تؤدي الإنتغرينات دورًا في دعم إشارات مستقبلات عوامل النمو السرطاني وترحيل الخلايا السرطانية والغزو المعتمِد على عوامل النمو السرطاني[36]. وتنظِّم الإنتغرينات أيضًا عملية الاستعمار في المواقع النقيلية عن طريق تسهيل بقاء الخلايا السرطانية المنتقلة في الدورة الدموية وغير الملتصقة بخلايا أخرى، حية طيلة فترة الانتقال.
وإضافة للإنتيغرينات، فإن هناك دورًا لبروتينات الكادهيرين (E-cadherin) أيضًا. تستخدم الخلايا النقيلية الكادهيرين في المواقع النقيلية كي تنفصل وتنتشر. يعزز هذا من بقاء الخلايا النقيلية ويمنع موت الخلايا المبرمج (Programmed cell death) الناتج من عوامل التأكسد. وبناءً عليه، فإن تثبيط الكادهيرين في خلايا السرطان النقيلية قد يكون له إمكانات علاجية ضد السرطان[37].
الخلايا السرطانية في الدورة الدموية
تُشكل الرحلة عبر الدورة الدموية بيئة قاسية لمعظم الخلايا السرطانية التي تتسلل إلى الأوعية الدموية. تحدد مكونات بيئة الدورة الدموية والخلايا المناعية الموجودة هناك والتفاعلات بين الخلايا السرطانية الدائرة في الدم، وهذه المكونات قدرة الخلايا السرطانية على البقاء وعلى الخروج لاحقًا إلى مواقع بعيدة[38]. في حين تنتشر معظم الخلايا السرطانية الدائرة في الدم بصورة خلايا فردية، تنتقل أنواع أخرى من الخلايا السرطانية على شكل مجموعات (عناقيد). تكون الخلايا الفردية أكثر عرضة للموت في أثناء هذه الرحلة، بينما تكون المجموعات العنقودية المنتشرة أكثر عرضة لتكوين الانبثاث. بالإضافة إلى الخلايا السرطانية الغازية، تحتوي المجموعات الخلوية السرطانية على خلايا اللُحمة والمكونات المناعية من البيئة المحيطة الأصلية التي تسهم في جعل التجمع الخلوي متباينًا وتعزز بقاءها[39]. تشارك الخلايا العدِلة في تكوين العناقيد وتثبط تنشيط كريات الدم البيضاء، ما يزيد من فرص بقاء الخلايا السرطانية الدائرة[40]. علاوة على ذلك، يؤدي التصاق الخلايا السرطانية الدائرة مع الصفائح الدموية إلى تكوين طبقة واقية من الصفائح الدموية حول الخلايا السرطانية، ما يمنع الكشف عن الخلايا السرطانية الدائرة بوساطة الخلايا المناعية وتوفر البنية اللازمة لتحمل الضغوط الفيزيائية للدورة الدموية[41].
التسريب: خروج الخلايا السرطانية من الأوعية الدموية
عندما تصل الخلايا السرطانية الدائرة في الدم للأعضاء البعيدة، مارةً عبر الشعيرات الدموية الصغيرة، فإنها تعلق بسبب ضيق المساحة، ما يؤدي إما إلى تمزق هذه الشعيرات الدموية أو يجبر الخلية على الخروج في عملية توصف بالتسريب {{التسريب (Extravasation): العملية التي تخرج فيها الخلايا، مثل السرطانية أو المناعية، من الأوعية الدموية أو اللمفاوية إلى الأنسجة المحيطة. وتحدث هذه العملية عادةً بعد دخول الخلايا إلى الأوعية الدموية، إذ تخترق الدورة الدموية منبعثة عن الورم الأساسي. وبمجرد أن تصبح الخلايا في الدم أو السائل اللمفاوي، قد تعلق بجدران الشعيرات الدموية، ثم تخرج إلى الأنسجة المجاورة.}}[42]. نظرًا إلى أن أعضاء في الجسم مثل الكبد والعظام تحتوي على أوعية دموية عالية النفاذية، فإن الخلايا السرطانية الدائرة تظهر معدلًا مرتفعًا من النقائل في هذه الأعضاء[43]. في الأعضاء الأخرى، تواجه الخلايا الخارجة حواجز صارمة وأغشية قاعدية تتطلب وجود عوامل وراثية وجزيئية متعددة حتى تتمكن من الانتقال عبر الغشاء. لذا فإن الانبثاث السرطاني عادة ما يتركز في الكبد والرئتين والعظام.
إن خروج الخلايا السرطانية عبر جدار الأوعية الدموية عملية معقدة تتضمن تفاعلات ارتباط بين الرُّبيطة {{الرُّبيطة: جزيء أو أيون أو مركب كبير يرتبط بمستقبِل أو بجزيء آخر، عادةً ما يكون بروتينًا، لتشكيل مركّب يمكن أن يُحفّز استجابة بيولوجية أو يسبب تفاعلًا كيميائيًّا.}} (Ligand)، والمستقبِل {{المستقبِل (Receptor): هو عادةً بروتين موجود على سطح الخلية أو داخلها، يرتبط بجزيئات معينة (مثل الرُّبيطات) لتحفيز استجابة بيولوجية.}}، والكيموكينات (Chemokines)، والخلايا غير الورمية الدائرة[44]. لبروتينات الإنتغرين، مرة أخرى، أدوار حيوية في تحديد المواقع التي يحدث فيها الخروج من الأوعية، وذلك عن طريق تسهيل بقاء الخلايا السرطانية الدائرة غير المرتبطة[45]. وكذلك، أوضحت الدراسات أن باستطاعة الخلايا السرطانية نفسها أن تحفّز حدوث النخر المبرمج (Necroptosis) للخلايا البطانية، ما يسهل على الخلايا النقيلية الخروج من مجرى الدم إلى الأعضاء البعيدة (الشكل 6).
[الشكل 7]
 خروج الخلايا السرطانية من الأوعية الدموية
خروج الخلايا السرطانية من الأوعية الدموية
حذف الصورة؟
سيؤدي هذا إلى نقل الصورة إلى سلة المهملات.
استعمار الأعضاء
تواجه الخلايا المنتشرة التي تخرج من الأوعية الدموية في الموقع المستهدف ظروفًا قاسية تجعل البقاء على قيد الحياة صعبًا[46]. تعمل مجموعة من العوامل السرطانية المفرزة وخلايا مشتقة من نخاع العظم على تحفيز تكوين الشبكة المحيطة بالورم، حيث تستعمر الخلايا السرطانية وتنمو[47]. بالإضافة إلى العوامل السرطانية المفرزة، تؤدّي الإكسوسومات {{الإكسوسوم (Exosomes): حويصلة صغيرة محاطة بغشاء، تُفرزها بعض الخلايا، وتحتوي على مواد مختلفة، مثل البروتينات والدهون والأحماض النووية الريبية (RNA). تؤدي الإكسوسومات دورًا في التواصل بين الخلايا ونقل الإشارات الجزيئية.}} دورًا في توجيه الخلايا الجذعية لنخاع العظم لتصبح نقيلية[48]. ففي سرطان البنكرياس، على سبيل المثال، أسهمت الإكسوسومات المفرزة بالبدء بتكوين شبكة محيطه بالورم في الكبد[49].
إضافة إلى ذلك، فإن تفاعلات الخلايا السرطانية مع الخلايا المستضيفة مهمة أيضًا للاستعمار المناسب. تتحكم الخلايا الكبدية في تراكم الخلايا النخاعية ما يؤدي إلى زيادة قابلية الكبد للاستعمار النقيلي. في نماذج الفئران المصابة بسرطان البنكرياس، وجد العلماء أن الخلايا الكبدية تحفز إشارات ستات 3 {{ستات 3: اختصار للناقل والمحفّز للتعبير الجيني 3 Signal Transducer and Activator of Transcription 3))، وهو عامل نسخ (Transcription factor) يُنشّط بوساطة السيتوكينات (وهي نوع من بروتينات الإشارة الصغيرة)، وعوامل النمو، وتتمثل وظيفته في تنظيم الاستجابات المناعية، وبقاء الخلايا، وتكاثرها وتمايزها.}} (STAT3) بوساطة إنترلوكين 6 {{إنترلوكين 6 (Interleukin-6, IL-6): سيتوكين يشارك في تنظيم الاستجابة المناعية والالتهابات، ويؤدي دورًا رئيسًا في الأمراض، مثل السرطان والاضطرابات المناعية الذاتية.}}، وتزيد من إفراز الأميلويد أ1 {{أميلويدأ1 (Amyloid A1): بروتين يُنتج في الكبد استجابة للالتهابات الحادة. يمكن أن يتراكم في الأنسجة المحيطة ويشكل رواسب أميلويد في حالات، مثل الأمراض الالتهابية المزمنة.}}، والأميلويد أ2 {{أميلويدأ2 (Amyloid A2): بروتين يشفّره جين SAA2، وهو عضو في عائلة الأميلويد المصلي (SAA) مثل أ1، ويُنتج في الكبد خلال استجابات المرحلة الحادة، ويمكن أن يُسهم في تكوين رواسب أميلويد في الأنسجة خلال الالتهابات المزمنة.}} في الدم. إن تثبيط الإشارات الخلوية من إنترلوكين 6 إلى ستات 3 والأميلويد يمنع إنشاء شبكة محيطة بالورم، ما يؤدي إلى إعاقة استعمار النقائل في الكبد[50].
[الشكل 8]
 حركة الخلايا السرطانية لاستعمار أعضاء جدد
حركة الخلايا السرطانية لاستعمار أعضاء جدد
حذف الصورة؟
سيؤدي هذا إلى نقل الصورة إلى سلة المهملات.
يُعدّ إنشاء شبكة أوعية دموية أمرًا ضروريًّا ليكون استعمار النقائل مناسبًا. فيدفع تكوين الأوعية الدموية قدرة بعض خلايا سرطان الثدي. على سبيل المثال، كي تسهم في إرسال النقائل البعيدة من خلال الإنتاج المفرط لبروتينات محددة، مثل سيرباين 2 {{سيرباين 2 (SERPINE2): مثبط لإنزيمات البروتياز، ويُسهم من خلال تأثيره في النشاط البروتيني في إعادة تشكيل الأنسجة والحد من تدمير الأوعية الدموية الموجودة أثناء تكوين أوعية جديدة. كما يدعم سيرابين 2 البيئة المحلية التي تسمح للأورام بالانتشار والنمو عن طريق تسهيل تكوين أوعية دموية جديدة تُغذي الأورام وتساعد في انتقال الخلايا السرطانية.}} ومثبِّط بروتياز السيرين {{مثبّط بروتياز السيرين (SLPI): يعمل على تثبيط البروتياز، وقد يظهر في الأنسجة التي تتطلب تنظيمًا دقيقًا للنمو الوعائي، كما يُشارك في تقليل الالتهابات أو تفاعل الأنسجة مع الأورام، ما قد يسهم في خلق بيئة تسمح بتطور النقائل.}}، وهي بروتينات لها أدوار أساسية في عملية تكوين الأوعية الدموية. إن التعبير الجيني المفرط (Overexpression) للجينين المشفّرين لهذين البروتينين مفضّل بشكل تفضيلي في مرضى سرطان الثدي البشري الذين يعانون من نقائل الرئة، ما يوحي بإمكانية حدوث تقدم انبثاثي بسبب هذين البروتينين[51].
سبات (كُمون) الخلايا السرطانية
يُعرّف سُبات أو كمون السرطان(Cancer dormancy) بأنه مرحلة يتوقف فيها تطور السرطان ونموّه، وتحدث هذه المرحلة في أثناء مرحلة تكون الورم الأساسي أو بعد الغزو إلى مواقع ثانوية[52]. يحدث الكمون الانبثاثي على وجه التحديد بسبب التأخر في تأقلم الخلايا السرطانية المنتشرة إلى المنافذ الثانوية الخاصة بها، فتستكين هذه الخلايا، وتقف عن النمو لفترات متغيرة، حتى تصبح الأمور مواتية لها لتتكاثر من جديد[53]. عند العديد من مرضى السرطان الذين اعتقدوا أنهم شفوا من مرضهم، تكون الخلايا السرطانية الكامنة موجودة بعد فترة طويلة من الإزالة الجذرية للورم الأساسي، ويُعتقد أنها مسؤولة عن الانتكاسات المتأخرة (Late relapses)[54]. هناك العديد من أوجه سبات السرطان، مثل الخمول الوعائي الذي يتحقق فيه التوازن شبه التام بين الخلايا السرطانية التي تنقسم وتلك التي تموت، والخمول الذي تسببه المناعة إذ تحافظ الخلايا المناعية على كتلة الورم عن طريق التخلص من الخلايا المنقسمة[55].
هناك تباين للرأي حول ما إذا كانت بيئة العضو المستهدف (Microenvironment) تُوجّه الخلايا السرطانية الدائرة للدخول في خمول. يعتقد بعض العلماء أن البيئة تؤثر في الخلايا الورمية وتدفعها إلى الخمول، بينما يعتقد علماء آخرون أن الأورام الأساسية تُشفّر مسبقًا إشارة تجبر الخلايا السرطانية على الخمول عند دخول الخلايا السرطانية الدائرة في الدم إلى بيئة المضيف[56] (الشكل 8).
[الشكل 9]
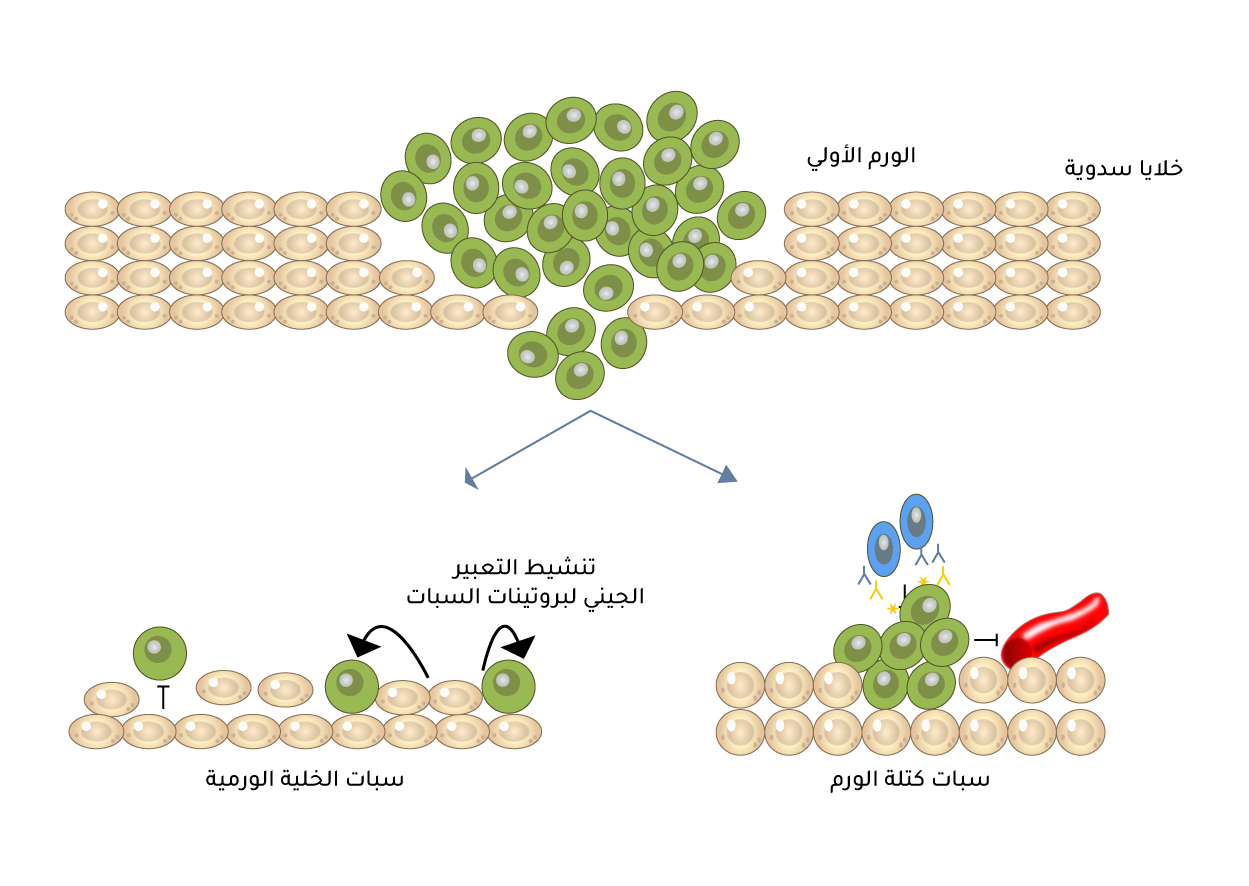 سبات الخلايا السرطانية
سبات الخلايا السرطانية
حذف الصورة؟
سيؤدي هذا إلى نقل الصورة إلى سلة المهملات.
آليات سبات الخلايا السرطانية
تُنظِّم التفاعلات المتبادل بين البيئة والآليات التي تتحكم بالتعبير الجيني بعملية سبات الخلايا السرطانية[57]. يوصف السبات أحادي الخلية بأنه حالة السكون القابلة للانعكاس التي تدخل فيها الخلية النقيلية استجابةً للمؤثرات المجهدة، مع تحفيز التعبير الجيني لبروتين مؤشر الانقسام البروتين Ki67{{بروتين :Ki67 هو بروتين يُنتَج في أنوية الخلايا التي تنقسم بسرعة، ويُستخدم عادةً بوصفه واسمًا حيويًّا (Biomarker) لتقييم تكاثر الخلايا ومعدل نمو الأورام في أبحاث السرطان.}}[58].
تحافظ الخلية السرطانية الكامنة على توازنها الأيضي من خلال تثبيط مسارين يُنَشّطا عادة في السرطان، وهما مسار راس الأيضي {{مسار راس الأيضي (RAS pathway): (أو مسار RAS – MEK – ERK – MAPK) مسار إشاري رئيس في الخلايا، ينظِّم العديد من العمليات الخلوية مثل النمو، والتكاثر، والتمايز، والبقاء.}}، ومسار فوسفاتيديل إينوسيتول 3-كيناز {{مسار فوسفاتيديل إينوسيتول 3-كيناز (PI3K-AKT-mTOR): مسار إشارة حيوي في الخلايا يُنظّم العديد من العمليات الأساسية مثل النمو، والبقاء، والتكاثر.}} وهما مساران يتحكمان في سبات الخلايا السرطانية[59].
إن العوامل التي تفرزها الشبكة المحيطة بالورم، مثل بروتينات التخليق العظمي {{بروتينات التخليق العظمي (Bone morphogenetic proteins): مجموعة من البروتينات الإشارية وعوامل النمو التي تنتمي إلى عائلة عامل النمو المحوِّل بيتا (TGF-β). تنظِّم هذه البروتينات مجموعة متنوعة من العمليات الخلوية، بما في ذلك التمايز الخلوي، والنمو، والتطوّر، وخاصة في تكوين العظام والغضاريف.}} المشتقة من خلايا اللحمة المتوسطة، تحول الخلايا السرطانية أيضًا نحو الكمون[60]. يُنشِّط بروتين BMP7 الجين المثبط للنقائل NDGR1، ما يؤدي إلى زيادة في تنشيط مسار بي 38 ماب كيناز {{مسار بي38 ماب كيناز (p38 MAPK): بروتين يُفعّل استجابة للعديد من المحفزات الخلوية، مثل الإجهاد الخلوي، الالتهابات، والإشارات الناتجة عن النمو. ينتمي بي38 إلى عائلة ماب كيناز التي تشمل أيضًا بروتينات، مثل إرك وجنك (JNK)، وتعمل على تنظيم مجموعة من العمليات الخلوية، مثل الاستجابة للإجهاد، والانقسام، والاستماتة الخلوية.}}، وزيادة في تعبيــر مثبط دورة الخلية، بروتين 21 {{بروتين 21 (p21): مثبّط للكيناز المعتمد على السيكلين (Cyclin-dependent kinase inhibitor)، وله دور أساسي في تنظيم دورة الخلية عن طريق منع الانتقال من مرحلة G1 إلى مرحلة S، ويعمل غالبًا على أنه مثبِّط للأورام، استجابةً لتلف الحمض النووي أو التوتر الخلوي.}}، وتعليقها[61].
إن التفاعلات الجزيئية بين الإشارات المحفزة للميتوجين {{الميتوجين (Mitogen): مادة كيميائية أو جزيء بروتيني يحفِّز انقسام الخلايا ويُفعّل مسارات الإشارة التي تعزز الانتقال من مرحلة G1 إلى مرحلة S في دورة الخلية.}} وإشارات الإجهاد ضرورية لتنظيم حالة سبات/تنشيط الخلايا السرطانية الانبثاثية. تنظم نسبة بروتين إرك 1/2 (ERK1/2) إلى بي 38 دورة الخلية، إذ إن المستويات العالية من نشاط بروتين إرك 1/2 تعزز الانقسام، بينما تعزز المستويات المرتفعة من بي 38 السبات. ويحفّز أيضًا زيادة نشاط بي 38 تنشيط استجابة البروتين غير المطوي (Unfolded protein response)، الذي يعزز تعليق دورة الخلية وبقاءها في حالة سبات[62]. لذا فمن الملاحظ أن تنشيط مسارات إشارات الإجهاد يضع الخلية في حالة سبات مستدامة.
السبات في العناقيد النقيلية
عندما يكون معدل تكاثر الخلايا داخل العنقود الورمي مساويًا لمعدل موت الخلايا المبرمج فيه يحدث السبات في العناقيد السرطانية النقيلية. وبناءً على ذلك، لا تتمدد العناقيد الورمية إلى نقائل مجهرية. يتحقق هذا التوازن من خلال إشارات الجينات المثبطة وتقييد إنتاج الأوعية الدموية و/ أو وجود بيئة مناعية نشطة[63]. يمكن تفعيل إشارات الجينات المثبطة من خلال تحفيز تعبير جين DEC2، وهو جين كابت للورم (Tumor suppressor gene). يحفّز عامل النمو المحوّل بيتا (Transforming growth factor-beta (TGF-β)) جين DEC2، ما يؤدي إلى تثبيط الكيناز المعتمد على السيكلين 4 (Cyclin-dependent kinase 4, CDK4) وتنشيط بروتين بي 27 {{بروتين بي 27 (p27): مثبط للكيناز المعتمد على السيكلين، يُنظّم دورة الخلية عن طريق تثبيط نشاط مجمّعات السيكلين-الكيناز المعتمدة على السيكلين (Cyclin-CDK)، خاصةً أثناء الانتقال من مرحلة G1 إلى مرحلة S، ويعمل على أنه مثبط للأورام.}}، ما يجبر الخلية على الدخول في حالة من السبات[64]. يؤدي منع تكوين الأوعية الدموية من خلال تنشيط بروتين ثرومبوسبوندين-1 (Thrombospondin-1)[65] أو من خلال تثبيط البروتينات المساندة، مثل بروتين الصدمة الحرارية 27 {{بروتين الصدمة الحرارية 27 (Heat shock protein 27): بروتين مساعد (شابرون Chaperone) جزيئي صغير، يساعد في حماية الخلايا من الإجهاد عن طريق مساعدته في طيّ البروتينات، واستقرارها، ومنع تكتّلها، خاصةً في ظروف، مثل الصدمة الحرارية أو الإجهاد الخلوي بشكل عام.}}، إلى دفع العناقيد النقيلية إلى حالة السبات[66].
إضافة إلى أن الجهاز المناعي أيضًا عامل رئيس في مكافحة تكاثر السرطان. فتُزيل الخلايا التائية والخلايا القاتلة الطبيعية، بالإضافة إلى الخلايا البلعمية، الخلايا النقيلية عن طريق التحلل الخلوي[67]. ومع ذلك، فبإمكان الخلايا السرطانية الكامنة أن تنتج كمية مستضدات قليلة للهروب من الجهاز المناعي، والذي قد يكون السبب وراء الانتكاسات بعد العلاج المناعي[68].
تنشيط الخلية الكامنة
تدخل الخلايا السرطانية في حالة سبات وانقسام بطيء عن طريق تثبيط مسارات الإشارة المختلفة، ومنها ما هو مدفوع ببروتين ونت {{بروتين ونت (Wnt): ينتمي إلى عائلة من بروتينات الإشارة التي تنظّم العديد من العمليات الخلوية، مثل تكاثر الخلايا، والتمايز، والهجرة، وتشكيل الأنسجة، وترتبط هذه البروتينات أيضًا بعمليات النمو، وبعدد من الأمراض، بما في ذلك السرطان.}}[69]. وإضافةً لذلك، تُظهر هذه الخلايا مستويات متزايدة من تحفيز إنتاج عوامل النسخ المرتبطة بالخلايا الجذعية SOX2 وSOX9، التي تسمح بنمو أورام جديدة إذا توفرت ظروف معينة. لتقليل قدرة الجهاز المناعي على تحديدها، تقلل الخلايا السرطانية الكامنة من التعبير عن الجزيئات القابلة للتعرف عليها من قبل الخلايا المناعية[70]، ما يسمح للخلايا السرطانية بالتهرب من الاستجابة المناعية إلى أن تسمح الظروف بتطور النقائل (الشكل 9). قد يؤدي الالتهاب المستمر في عضو المضيف إلى تحويل الخلايا السرطانية الكامنة إلى نقائل عدوانية[71]. وتجدر الإشارة إلى أن التحول من السبات إلى التنشيط مرتبط بالمستويات العالية من إرك 1/2 بالنسبة إلى بي 38.
[الشكل 10]
 إعادة تنشيط الخلية السرطانية الكامنة
إعادة تنشيط الخلية السرطانية الكامنة
حذف الصورة؟
سيؤدي هذا إلى نقل الصورة إلى سلة المهملات.
مرونة الخلايا السرطانية وتطور الورم
تسهم مرونة الخلايا السرطانية في ظهور خصائص مقاومة العلاج (Chemoresistance) وتطور الورم الخبيث. إن مرونة الخلايا السرطانية تمنحها القدرة على التحول ديناميكيًّا بين الحالة المتمايزة ذات الإمكانية المحدودة لتكوّن الورم، وحالة الخلية غير المتمايزة أو الشبيهة بالخلايا الجذعية السرطانية، المسؤولة عن نمو الورم على المدى الطويل. ومع ذلك، يظل الباحثون متفائلين بإمكانية استغلال مرونة الخلايا السرطانية لأغراض علاجية. فقد أجبر بعض العلماء الخلايا السرطانية الثديّة المشتقة من التحول الطلائي إلى اللحمة المتوسطية على التحول التفاضلي إلى خلايا دهنية وظيفية (Functional adipocytes) عن طريق استخدام العلاج المزدوج الذي يجمع بين مثبطات بروين ميك (MEK) ودواء روزيغليتازون (Rosiglitazone) الذي يُستخدم لعلاج مرضى السكري من النوع الثاني (Type 2 Diabetes Mellitus)، ما يثبط بذلك العملية النقيلية[72].
يمكن لعمليات الفحص الحيوي على نطاق الجينوم بأكمله تحديد منظِّمات جديدة للاستعمار النقيلي. حددت الدراسات الحيوية العديد من الجينات التي يؤدي تعطيلها إلى تعديل قدرة الخلايا السرطانية على إنشاء نقائل[73]. في كثير من الأحيان، تعمل الخلايا الجذعية البطانية الداخلية (Endothelial stem cells) بوصفها سلائف للخلايا البطانية[74]. تُعدّ هذه الخلايا عن عامل النسخ SOX18، وبناءً على ذلك، فهي لا تتأثر بالعلاجات التي تستهدف عامل النمو البطاني الوعائي (Vascular endothelial growth factor).
في العديد من الحالات، تستخدم الستيرويدات القشرية (Corticosteroids) لعلاج المرضى الذين يعانون من مضاعفات متعلقة بالسرطان. يبدأ تقدم سرطان الثدي بزيادة مستويات هرمون الإجهاد والستيرويدات القشرية، ما يؤدي بعد ذلك إلى تنشيط مستقبلات الستيرويدات القشرية في الموقع الثانوي وتعزيز استعمار السرطان وتقليل معدلات البقاء على قيد الحياة[75]، فيؤدي هذا إلى توخي الحذر عند استخدام الستيرويدات القشرية عند علاج مرضى السرطان.
على الرغم من الفعالية الواضحة للعلاج الكيميائي في علاج سرطان الثدي، فقد ثبت أن بعض العلاجات تُظهر تأثيرات انبثاثية[76]. يحفِّز دوائي باكليتاكسيل (Paclitaxel) ودوكسوروبيسين (Doxorubicin) إنتاج حويصلات خارج الخلايا مشتقة من الورم في نماذج سرطان الثدي المقاوم للعلاج الكيميائي لدى الفئران[77]. تسهل هذه الحويصلات استعمار الأورام في المواقع النقيلية بالرئة[78].
إسهام الميكروبيوم في الانبثاث
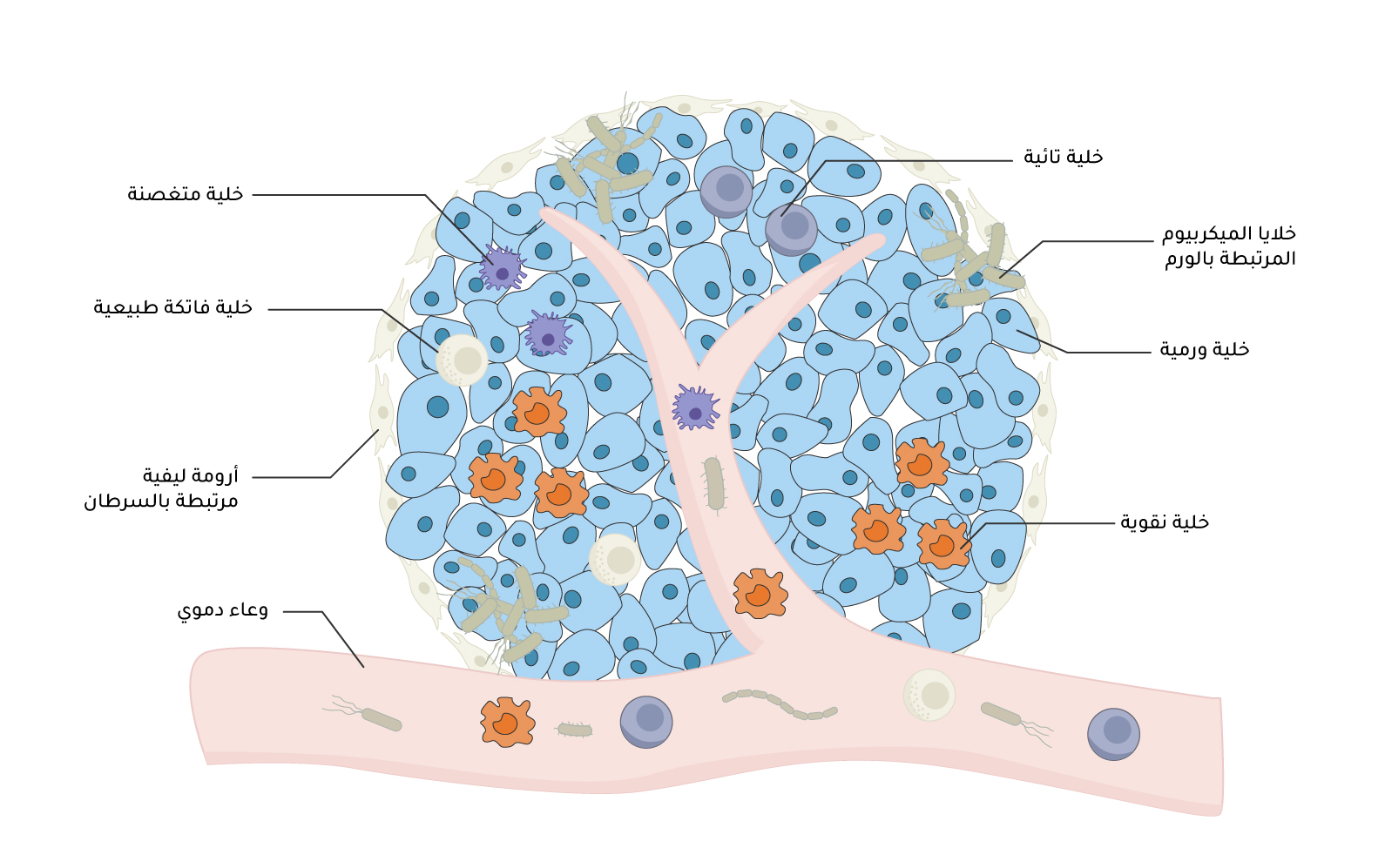
يشير مصطلح ميكروبيوم الورم (Tumor microbiome) إلى مجتمع الكائنات الحية الدقيقة، بما في ذلك البكتيريا والفطريات والفيروسات والميكروبات الأخرى، التي تعيش داخل الورم وحوله (الشكل 10). يمكن أن تؤثر هذه الميكروبات في تطور الورم وتقدمه واستجابته للعلاجات. نشأ المفهوم من اكتشاف مجموعات بكتيرية داخل الأورام، وفي حين لا تزال العلاقة الدقيقة بين ميكروبيوم الورم والنتائج السريرية قيد الدراسة، تشير بعض الدراسات إلى أن بعض الميكروبات قد تكون مرتبطة بأنواع معينة من السرطان[79]. على سبيل المثال، قد تستعمر الميكروبات الأورام بشكل تفضيلي مع شبكات الأوعية الدموية الغنية والبيئات المنخفضة الأكسجين، وهو أمر شائع في العديد من أنواع السرطان. إن البكتيريا اللاهوائية (Anaerobic bacteria) أو اللاهوائية الاختيارية Facultative anaerobic) bacteria)، على وجه التحديد، تنجو بقوة في البيئات المحيطة بالورم منخفضة الأكسجين[80]. يمكن أن تؤثر هذه البكتيريا أيضًا في فعالية العلاج الكيميائي عن طريق استقلاب أدوية العلاج الكيميائي أو تعديلها، ما يؤدي إلى التأثير في كيفية استجابة الأورام للعلاج[81].
هناك العديد من الآليات التي تفسر تأثير ميكروبيوم الورم في النقائل:
أ- تعديل الاستجابة المناعية: يمكن أن تؤثر الكائنات الحية الدقيقة داخل الأورام في الاستجابة المناعية، فقد تعمل بعض البكتيريا على تعديل البيئة المحيطة بالورم من خلال التأثير في الخلايا المناعية، مثل الخلايا البلعمية أو الخلايا التائية، والتي بدورها يمكن أن تؤثر في قدرة الخلايا السرطانية على الانتشار.
ب- تغيير في البيئة المحيطة بالورم: يمكن أن يؤثر الميكروبيوم في الحالة الأيضية والالتهابية للورم، ما يخلق ظروفًا تدعم هجرة الخلايا السرطانية وغزوها. على سبيل المثال، قد تساعد البكتيريا في تغيير المصفوفة خارج الخلية أو زيادة نفاذية الأوعية الدموية، ما يسهل حركة الخلايا السرطانية إلى مجرى الدم أو الجهاز اللمفاوي.
ت- العوامل المرتبطة بالنقائل: لقد ثبت أن بعض الأنواع الميكروبية تنتج مستقلِبات (Metabolites) أو بروتينات تعزّز عمليات محددة، مثل تكوين الأوعية الدموية أو إعادة تشكيل الأنسجة، وكلاهما مهم للانتشار النقيلي. قد تؤثر البكتيريا أيضًا في التعبير عن الجينات المرتبطة بعدوانية الورم، مثل تلك التي تشارك في التحول الطلائي اللُحمي المتوسط، وهي عملية رئيسة في النقائل.
ث- تأثير البكتيريا في مقاومة الأدوية: قد يؤثر الميكروبيوم أيضًا في كيفية استجابة الخلايا السرطانية للعلاجات، ما قد يسهم في تطوير مقاومة العلاج الكيميائي أو العلاج المناعي، ليسهل بذلك بشكل غير مباشر انتشار النقائل.
[الشكل 11]
 استراتيجيات علاجية
استراتيجيات علاجية
حذف الصورة؟
سيؤدي هذا إلى نقل الصورة إلى سلة المهملات.
إن علاج سرطان النقائل له تحديات وعقبات كبيرة بسبب نقص التجارب السريرية التي تستهدف النقائل وقلة المعرفة الدقيقة بالأسس البيولوجية التي تحكُم عملية الانبثاث[82]. إضافة إلى أن تصميم علاجات موجهة لخلايا سرطان النقائل تحتاج مراعاة الاختلافات الجينية والنمطية بين الخلايا الأصلية والنقيلية المتجولة[83]. حتى نهاية عام 2024، دائمًا ما يرتبط تشخيص سرطان النقائل بأنه مرض مميت. وعلى الرغم من وجود بعض النتائج التي تظهر منع النقائل بشكل سريري في السابق، إلا أن تطوير الأدوية قد تعثر بسبب ضعف تصميم التجارب والاستراتيجيات العلاجية الملائمة. ومع ذلك، أدى التقدّم في العلاج المناعي إلى تحسين معدلات البقاء على قيد الحياة والنتائج الصحية للمرضى المصابين بالورم الميلاني النقيلي[84]. إضافة إلى ذلك، أدى تطوير مثبِّطات مستقبِلات الأندروجين (Androgen receptors inhibitors) الجديدة إلى إطالة عمر مرضى سرطان البروستاتا النقيلي[85]. ولكن، فشلت المتابعات طويلة المدى في إثبات تناسق في فوائد البقاء على قيد الحياة لمرضى سرطان الثدي النقيلي[86].
لتطوير استراتيجيات علاجية ملائمة، لجأ العلماء إلى دراسة واستهداف مسارات محددة في سلسلة النقائل[87]. يمكن استهداف انغراس الخلايا السرطانية عن طريق تثبيط التفاعلات داخل الورم الأصلي، وأيضًا عن طريق ضرب التفاعل بين الخلايا من خلال جزيئات الالتصاق في المصفوفة خارج الخلية، ومنع إطلاق البروتينات المحلِّلة للبروتين، وتثبيط عمليتي التحول الطلائي اللحمي المتوسط والغزو الوعائي. ومع ذلك، هناك العديد من المتغيرات التي تحد من فعالية هذه التدخلات الاستباقية. على سبيل المثال، من الممكن أن تكون الخلايا السرطانية قد انتقلت بالفعل إلى الجهاز الدوري أو حتى استعمرت موقعًا بعيدًا، وقت تشخيص النقائل[88]. لذلك، يبدو أن استهداف الاستعمار النقيلي اللاحق هو أكثر استراتيجية علاجية منطقية وفعالية، إذ إنها ترتبط بشكل أساسي بالمشهد السريري للمريض.
تثبط مثبطات الانبثاث نمو السرطان وتكاثره في موقع النقيلة دون التأثير في الورم الأولي[89]. وهي تستهدف المسارات والبروتينات المسببة للأورام السرطانية التي تشارك في الغزو والاستعمار النقيلي في نهاية المطاف. على سبيل المثال، بروتين 8 المرتبط بالكيناز أ (A-kinase anchor protein 8) عامل تنظيمي يثبط التحول الطلائي اللحمي المتوسط، ويثبط انتشار سرطان الثدي[90]. وفي الخلايا شديدة الانتشار، عادةً ما تثبّط عمليات تنظيم تكوين مثبطات النقائل في الخلايا السرطانية الأولية[91]، ما يجعل الخلية قادرة على الغزو. في أوائل الألفية الثانية، حدّد الباحثون عددًا كبيرًا من مثبطات النقائل. على وجه الخصوص، وجدوا جزيئات من الحمض النووي الريبي الميكروي {{الحمض النووي الريبيالميكروي MicroRNAs)): جزيء قصير السلسلة من الحمض النووي الريبي، لا يُشفّر أي ناتج بيولوجي، ولكنه يُنظِّم التعبير الجيني عن طريق الارتباط بجزيئات الحمض النووي الريبي المرسال (Messenger RNA)، ما يؤدي إلى تثبيط ترجمتها إلى بروتينات أو تحفيز تحللها.}} التي تسهم في تثبيط تعبير الجينات الورمية وإشارات الورم الخبيث، فهي تُعدّ أهدافًا محتملة لرصد الورم الخبيث وتدميره[92].
درس العلماء كذلك سبات الخلايا بوصفه هدفًا محتملًا لمنع استعمار النقائل، فاقترح بعضهم علاجات تساعد في الحفاظ على الحالة الكامنة[93]، في حين صمّم آخرون علاجات مشتركة تستهدف الخلايا السرطانية مباشرةً في طور السبات (G0) من دورة حياة الخلية. علاوةً على ذلك، طُوِّرت أجسام مضادة وحيدة النسيلة {{الأجسام المضادة وحيدة النسيلة (Monoclonal antibodies): نوع من المنتجات البيوتكنولوجية يُحضّر مخبريًا عن طريق توليد نسخ متطابقة (نسائلclones ) من خلية مناعية واحدة، تُعرف بالخلايا البائية (B cells)، وهي الخلايا التي تنتج جسمًا مضادًا محددًا يستهدف مستضدًا معينًا.}} لاستهداف الخلايا السرطانية الفردية في هذه المرحلة[94].
يظل الدماغ موقعًا خاصًا للنقائل، إذ يتوفّر به الملاذ الآمن للخلايا المستعمرة بسبب وجود الحاجز الدموي الدماغي (Blood-brain barrier). في حين يسمح الحاجز الدموي الدماغي بعبور بعض الخلايا السرطانية من خلاله، ويمنع مرور العوامل العلاجية[95]. لذلك، يجتهد العلماء لاختبار العوامل والمواد العلاجية التي يُعرف عنها قدرتها على عبور الحاجز الدموي الدماغي في حالات نقائل الدماغ، ويركّزون أيضًا على تصميم أدوية جديدة لها القدرة على عبور هذا الحاجز[96]. بشكل عام، تُشكّل النقائل تحديًا معقدًا يتطلب استخدام أكثر من استراتيجية علاجية واحدة لكبحها. لذلك، يبدو أن تبني نموذج العلاج المشترك واستهداف مسارات متعددة في وقت واحد هو المفتاح لمواجهة التغيرات الجينية والنمطية المهمة التي تقدمها خلايا سرطان النقائل[97].
المراجع
Yeh C. & Ramaswamy S. “Mechanisms of cancer cell dormancy—another hallmark of cancer?.” Cancer research. vol. 75, no. 23 (2015). pp. 5014-5122.
Aceto, N. et al. “Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis.” Cell. vol. 158, no. 5 (2014). pp .1110-1122.
Aguirre-Ghiso, Julio A. & Maria Soledad Sosa. “Emerging topics on disseminated cancer cell dormancy and the paradigm of metastasis.” Annual Review of Cancer Biology. vol. 2 (2018). pp. 377-393.
Aguirre-Ghiso, Julio A. et al. “Urokinase receptor and fibronectin regulate the ERK(MAPK) to. p. 38 (MAPK) activity ratios that determine carcinoma cell proliferation or dormancy in vivo.” Molecular Biology of the Cell. vol. 12, no. 4 (2001). pp. 863-879.
Albrengues, Jean et al. “Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice.” Science. vol. 361, no. 6409 (2018).
Alexender, Ring et al. “Biology, vulnerabilities and clinical applications of circulating tumour cells.” Nature Reviews Cancer. vol. 23, no. 2 (2023). pp. 95-111.
Bakhoum, Samuel F. et al. “Chromosomal instability drives metastasis through a cytosolic DNA response.” Nature. vol. 55, no. 76893 (2018). pp. 467-72.
Bockhorn, Maximilian. Rakesh K. Jain & Lance L. Munn. “Active versus passive mechanisms in metastasis: do cancer cells crawl into vessels, or are they pushed?.” The lancet oncology. vol. 8, no. 5 (2007). pp .444-448.
Cheung, Kevin J. & Andrew J. Ewald. “A collective route to metastasis: Seeding by tumor cell clusters.” Science. vol. 352, no. 6282 (2016). pp. 167-169.
Clark, Andrew G. & Danijela Vignjevic. “Modes of cancer cell invasion and the role of the microenvironment.” Current Opinion in Cell Biology. vol. 36 (2015). pp. 13-22.
Costa-Silva, Bruno et al. “Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver.” Nature Cell Biology. vol. 17, no. 6 (2015). pp. 816-826.
De Craene, Bram & Geert Berx. “Regulatory networks defining EMT during cancer initiation and progression.” Nature Reviews Cancer. vol. 13, no. 2 (2013). pp. 97-110.
Denais, Celine M. et al. “Nuclear envelope rupture and repair during cancer cell migration.” Science. vol. 352, no. 6283 (2016). pp. 8-353.
Donovan, Prudence et al. “Endovascular progenitors infiltrate melanomas and differentiate towards a variety of vascular beds promoting tumor metastasis.” Nature Communications. vol. 10, no. 1 )2019). p. 18.
Douglas, Hanahan & Robert A. Weinberg. “Hallmarks of cancer: the next generation.” Cell. vol. 144, no. 5 (2011). pp .646-674.
Ecker, Ecker L. et al. “Age-Related Changes in HAPLN1 Increase Lymphatic Permeability and Affect Routes of Melanoma Metastasis.” Cancer Discovery. vol. 9, no. 1 (2019). pp. 82-95.
Erdogan, Begum et al. “Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin.” Journal of Cell Biology. vol. 216, no. 11 (2017). pp. 3799-3816.
Fares, Jawad et al. “Diagnostic Clinical Trials in Breast Cancer Brain Metastases: Barriers and Innovations.” Clinical Breast Cancer. vol. 19, no. 6 (2019). pp. 383-391.
________ et al. “Current state of clinical trials in breast cancer brain metastases.” Neuro-Oncology Practice. vol. 6, no. 5 (2019). pp. 392-401.
________, Mohamad Y. Fares & Youssef Fares. “Immune checkpoint inhibitors: Advances and impact in neuro-oncology.” Surgical Neurology International. vol. 10, no. 9 (2019).
________ et al. “Landscape of combination therapy trials in breast cancer brain metastasis.” International Journal of Cancer. vol. 147, no. 7 (2020). pp. 1939-1952.
Fidler, Isaiah J. “Metastasis: quantitative analysis of distribution and fate of tumor emboli labeled with 125 I-5-iodo-2'-deoxyuridine.” Journal of the National Cancer Institute. vol. 45, no. 4) 1970). pp. 773-782.
Fouad, Yousef Ahmed & Carmen Aanei. “Revisiting the hallmarks of cancer.” American Journal of Cancer Research. vol. 7, no. 5 (2017). pp. 1016-1036.
Gay, Lindsey J. & Barbara Felding-Habermann. “Contribution of platelets to tumour metastasis.” Nature Reviews Cancer. vol. 11, no. 2 (2011). pp. 123-134.
Ghajar, Cyrus M. et al. “The perivascular niche regulates breast tumour dormancy.” Nature Cell Biology. vol. 15, no. 7 (2013). pp. 807-817.
Giancotti, Filippo G. “Mechanisms governing metastatic dormancy and reactivation.” Cell. vol. 155, no. 4 (2013). pp. 750-7.64
Gomis, Roger R. & S. Gawrzak. “Tumor cell dormancy.” Molecular Oncology. vol. 11, no. 1 (2017). pp. 62-78.
Goss, Paul E. & Ann F. Chambers. “Does tumour dormancy offer a therapeutic target?.” Nature Reviews Cancer. vol. 10, no. 12 (2010). pp. 871-877.
H., Endo & Inoue M. “Dormancy in cancer.” Cancer Science. vol. 110, no. 2 (2019). pp. 474-480.
Hamidi, Hellye & Johanna Ivaska. “Every step of the way: integrins in cancer progression and metastasis.” Nature Reviews Cancer. vol. 18, no. 9 (2018). pp.533 -548.
Helmink, Beth A. “The microbiome, cancer, and cancer therapy.” Nature Medicine. vol. 25, no. 3 (2019). pp.377 -388.
Hodi, F. Stephen et al. “Improved survival with ipilimumab in patients with metastatic melanoma.” TheNew England Journal of Medicine. vol. 363, no. 8 (2010). pp. 711-723.
Hu, Xiaohui et al. “The RNA-binding protein AKAP8 suppresses tumor metastasis by antagonizing EMT-associated alternative splicing.” Nature Communications. vol. 11, no. 1 (2020). p. 486.
Ishay-Ronen, Dana et al. “Gain Fat-Lose Metastasis: Converting Invasive Breast Cancer Cells into Adipocytes Inhibits Cancer Metastasis.” Cancer Cell. vol. 35, no. 1 (2019). pp .17-32.
Keklikoglou, Ioanna et al. “Chemotherapy elicits pro-metastatic extracellular vesicles in breast cancer models.” Nature Cell Biology. vol. 21, no. 2 (2019). pp. 190-202.
Khan, Imran & Patricia S. Steeg. “Metastasis suppressors: functional pathways.” Laboratory Investigation. Vol. 98, no. 2) 2018). pp. 198-210.
Knott, Simon R. V. et al. “Asparagine bioavailability governs metastasis in a model of breast cancer.” Nature. vol. 554, no. 7692) 2018). pp. 378-81.
Kobayashi, A. et al. “Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone.” Journal of Experimental Medicine. vol. 208, no. 13) 2011(. pp. 2641-2655.
Lambert, Arthur W., Dwikar R. Pattabiraman & Robert A. Weinberg. “Emerging Biological Principles of Metastasis.” Cell. vol. 168, no. 4 (2017). pp. 670-91.
Lamouille, Samy, Jian Xu & Rick Derynck. “Molecular mechanisms of epithelial-mesenchymal transition.” Nature Reviews Molecular Cell Biology. vol. 15, no. 3 (2014). pp. 178-196.
Leach, James, Jennifer P. Morton & Owen J. Sansom. “Neutrophils: Homing in on the myeloid mechanisms of metastasis.” Molecular Immunology. vol. 110 (2019). pp. 69-76.
Lee, Jae W. et al. “Hepatocytes direct the formation of a pro-metastatic niche in the liver.” Nature. vol. 567, no. 7747) 2019). pp. 249-252.
Marshall, Jean-Ckaude et al. “Effect of inhibition of the lysophosphatidic acid receptor 1 on metastasis and metastatic dormancy in breast cancer.” Journal of the National Cancer Institute. vol. 104, no. 17) 2012). pp. 1306-1319.
Massague, Joan & Anna C. Obenauf. “Metastatic colonization by circulating tumour cells.” Nature. vol. 529, no. 7586) 2016). pp. 298-306.
________ & Karuna Ganesh. “Metastasis-Initiating Cells and Ecosystems.” Cancer Discovery. vol. 11, no. 4) 2021). pp. 971-94.
Nieto, Maria Angela et al. “EMT: 2016.” cell. vol. 166, no. 1 (2016). pp. 21-45.
Obradovic, Milan M. S. et al. “Glucocorticoids promote breast cancer metastasis. ” Nature. vol. 567, no. 7749 (2019). pp. 540-544.
P., Bragado et al. “TGF-β2 dictates disseminated tumour cell fate in target organs through TGF-β-RIII and p38α/β signalling.” Nature cell biology. vol. 15, no. 11 (2013). pp. 1351-1361.
Padmanaban, Veena et al. “E-cadherin is required for metastasis in multiple models of breast cancer.” Nature. vol. 573, no. 7774) 2019). pp. 439-444.
Panebianco, Concetta, Angelo Andriulli & Valerio Pazienza. “Pharmacomicrobiomics: exploiting the drug-microbiota interactions in anticancer therapies.” Microbiome. vol. 6, no. 1 (2018). p. 92.
Pantel, Klaus & Michael R. Speicher. “The biology of circulating tumor cells.” Oncogene. vol. 35, no. 10 (2016). pp. 1216-1224.
Pastushenko, Levgenia. et al. “Identification of the tumour transition states occurring during EMT.” Nature. vol. 556, no. 7702 (2018). pp. 463-468.
Peinado, Héctor, Simon Lavotshkin & David Lyden. “The secreted factors responsible for pre-metastatic niche formation: old sayings and new thoughts.” Seminars in cancer biology. vol. 21, no. 2 (2011).
Pushalkar, Smruti et al. “The Pancreatic Cancer Microbiome Promotes Oncogenesis by Induction of Innate and Adaptive Immune Suppression.” Cancer Discovery. vol. 8, no. 4 (2018). pp. 403-416.
Quail, Daniela F. & Johanna A Joyce. “Microenvironmental regulation of tumor progression and metastasis.” Natture Medicine. vol. 19, no. 11 (2013). pp. 1423-1437.
Rankin, Erinn B. & Amato J. Giaccia. “Hypoxic control of metastasis.” Science. vol. 352, no. 6282 (2016). pp. 175-180.
Reymond, Nicolas, Bárbara Borda d'Agua & Anne J. Ridley. “Crossing the endothelial barrier during metastasis.” Nature Reviews Cancer. vol. 13, no. 12 (2013). pp. 858-870.
Scher, Howard I. et al. “Increased survival with enzalutamide in prostate cancer after chemotherapy.” theNew England Journal of Medicine. vol. 367, no. 13 (2012). pp. 1187-1197.
Sonnenschein, Carlos & Ana M. Soto. “Carcinogenesis explained within the context of a theory of organisms.” Progress in Biophysics and Molecular Biology. vol. 122, no. 1 (2016). pp. 70-6.
Sosa, Maria Soledad, Emily Bernstein & Julio A. Aguirre-Ghiso. “Epigenetic regulation of cancer dormancy as a plasticity mechanism for metastasis initiation.” in: Yuzhuo Wang, Francesco Crea (eds.). Tumor Dormancy and Recurrence. Cham: Humana Press, 2017.
Straume, O. et al. “Suppression of heat shock protein 27 induces long-term dormancy in human breast cancer.” Proceedings of the National Academy of Sciences of the United States of America. vol. 109, no. 22 (2012). pp. 8699-8704.
Tabassum, Doris P. & Kornelia Polyak. “Tumorigenesis: it takes a village.” Nature Reviews Cancer. vol. 15, no. 8 (2015). pp. 473-483.
Tevaarwerk, Amye J. et al. “Survival in patients with metastatic recurrent breast cancer after adjuvant chemotherapy: little evidence of improvement over the past 30 years.” Cancer. vol. 119. no. 6 (2013). pp. 1140-1148.
Ulasov, Ilya et al. “Editing cytoprotective autophagy in glioma: an unfulfilled potential for therapy.” Trends in Molecular Medicine. vol. 26, no. 3 (2020). pp. 252-262.
Valastyan, Scott & Robert A. Weinberg. “Tumor metastasis: molecular insights and evolving paradigms.” Cell. vol. 147, no. 2 (2011). pp. 275-292.
Van der Weyden, Louise et al. “Genome-wide in vivo screen identifies novel host regulators of metastatic colonization.” Nature. vol. 541, no. 7636 (2017). pp. 233-336.
Wagenblast, Elvin et al. “A model of breast cancer heterogeneity reveals vascular mimicry as a driver of metastasis.” Nature. vol. 520, no. 7547 (2015). pp. 358-362.
Ye, Xin & Robert A. Weinberg. “Epithelial-Mesenchymal Plasticity: A Central Regulator of Cancer Progression.” Trends Cell Biology. vol. 25, no. 11 (2015). pp. 86-675.
Yoko, Katsuno, Lamouille Samy &Derynck Rik “TGF-beta signaling and epithelial-mesenchymal transition in cancer progression.” Current Opinion in Oncology. vol. 25, no. 1 (2013). pp. 76-84.
Zervantonakis, Ioannis K. et al. “Three-dimensional microfluidic model for tumor cell intravasation and endothelial barrier function.” Proceedings of the National Academy of Sciences of the United States of America. vol. 109, no. 34 (2012). pp. 13515-13520.
[1]Arthur W. Lambert, Dwikar R. Pattabiraman & Robert A. Weinberg, “Emerging Biological Principles of Metastasis,” Cell, vol. 168, no. 4) 2017), pp. 670-691.
[2] Carlos Sonnenschein & Ana M. Soto, “Carcinogenesis explained within the context of a theory of organisms,” Progress in Biophysics and Molecular Biology, vol. 122, no. 1 (2016), pp. 70-76.
[3] Lambert, Pattabiraman & Weinberg, pp. 670-91.
[4] Isaiah J. Fidler, “Metastasis: quantitative analysis of distribution and fate of tumor emboli labeled with 125 I-5-iodo-2'-deoxyuridine,” Journal of the National Cancer Institute, vol. 45, no. 4) 1970), pp. 773-782.
[5] Ring Alexender et al., “Biology, vulnerabilities and clinical applications of circulating tumour cells,” Nature Reviews Cancer, vol. 23, no. 2 (2023), pp. 95-111.
[6]Daniela F Quail & Johanna A Joyce, “Microenvironmental regulation of tumor progression and metastasis,” Nature Medicine, vol. 19, no. 11 (2013), pp. 1423-1437.
[7] Joan Massagué & Karuna Ganesh, “Metastasis-Initiating Cells and Ecosystems,” Cancer Discovery, vol. 1, no. 4 )2021), pp. 971-994.
[8] Ibid.
[9] Lambert, Pattabiraman & Weinberg, pp. 670-91.
[10] Samuel F. Bakhoum et al., “Chromosomal instability drives metastasis through a cytosolic DNA response,” Nature, vol .553, no. 7689 (2018), pp. 467-472.
[11] Doris P. Tabassum & Kornelia Polyak, “Tumorigenesis: it takes a village,” Nature Reviews Cancer, vol. 15, no. 8 (2015), pp. 473-483.
[12] Andrew G Clark & Danijela Vignjevic, “Modes of cancer cell invasion and the role of the microenvironment,” Current Opinion in Cell Biology, vol. 36 (2015), pp. 13-22.
[13] Kevin J. Cheung & Andrew J. Ewald, “A collective route to metastasis: Seeding by tumor cell clusters,” Scienc, vol. 352, no. 6282 (2016), pp. 167-169.
[14] Yousef Ahmed Fouad & Carmen Aanei, “Revisiting the hallmarks of cancer,” American Journal of Cancer Research, vol. 7, no. 5 (2017), pp. 1016-1036.
[15] Hanahan Douglas& Robert A. Weinberg, “Hallmarks of cancer: the next generation,” Cell, vol. 144, no. 5 (2011), pp .646-674.
[16] Xin Ye & Robert A. Weinberg, “Epithelial-Mesenchymal Plasticity: A Central Regulator of Cancer Progression,” Trends in Cell Biology, vol. 25, no. 11 (2015), pp. 675-686.
[17] Simon R. V. Knott et al., “Asparagine bioavailability governs metastasis in a model ofbreast cance,” Nature,vol. 554, no. 7692 )2018), pp. 378-381.
[18] Erinn B. Rankin& Amato J. Giaccia, “Hypoxic control of metastasis,” Science, vol .352, no. 6282 )2016), pp. 175-180.
[19] Samy Lamouille, Jian Xu & Rik Derynck, “Molecular mechanisms of epithelial-mesenchymal transition,” Nature Reviews Molecular Cell Biology, vol. 15, no. 3 (2014), pp. 178-196.
[20] Scott Valastyan & Robert A. Weinberg, “Tumor metastasis: molecular insights and evolving paradigms,” Cell, vol. 147, no.2 (2011), pp. 275-292.
[21] Brett L. Ecker et al., “Age-Related Changes in HAPLN1 Increase Lymphatic Permeability and Affect Routes of Melanoma Metastasis,” Cancer Discovery, vol. 9, no. 1 (2019), pp. 82-95.
[22] Maria Angela Nieto et al., ”EMT: 2016,” cell, vol. 166, no. 1(2016), pp .21-45.
[23] Ibid.
[24] Katsuno Yoko, Lamouille Samy & Derynck Rik, “TGF-beta signaling and epithelial-mesenchymal transition in cancer progression,” Current Opinion in Oncology, vol. 25, no. 1 (2013), pp. 76-84.
[25] Bram De Craene & Geert Berx, “Regulatory networks defining EMT during cancer initiation and progression,” Nature Reviews Cancer, vol. 13, no. 2 (2013), pp. 97-110.
[26] Levgenia Pastushenko et al., “Identification of the tumour transition states occurring during EMT,” Nature, vol. 556, no. 7702 (2018), pp. 463-468.
[27] Ibid.
[28] Ibid.
[29] Begum Erdogan et al., “Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin,” Journal of Cell Biology, vol. 216, no. 11) 2017), pp. 3799-3816.
[30] Fouad & Aanei, pp. 1016-1036.
[31] Maximilian Bockhorn, Rakesh K. Jain& Lance L. Munn, “Active versus passive mechanisms in metastasis: do cancer cells crawl into vessels, or are they pushed?,” The lancet Oncology, vol. 8, no. 5 )2007), pp. 444-448.
[32] Ioannis K. Zervantonakis et al., “Three-dimensional microfluidic model for tumor cell intravasation and endothelial barrier function,” Proceedings of the National Academy of Sciences of the United States of America, vol. 109, no. 34) 2012), pp. 13515-13520.
[33] Celine M. Denais et al., “Nuclear envelope rupture and repair during cancer cell migration,” Science, vol. 352, no. 6283) 2016), pp. 8-353.
[34] Ibid.
[35] Hellye Hamidi & Johanna Ivaska, “Every step of the way: integrins in cancer progression and metastasis,” Nature Reviews Cancer, vol. 18, no. 9) 2018), pp. 533-548.
[36] Ibid.
[37]Veena Padmanaban et al., “E-cadherin is required for metastasis in multiple models of breast cancer,” Nature, vol. 573, no. 7774) 2019), pp .439-444.
[38] Klaus Pantel & Michael R. Speicher, “The biology of circulating tumor cells,” Oncogene, vol. 35, no. 10 (2016), pp. 1216-1224.
[39] N. Aceto et al., “Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis,” Cell, vol. 158, no. 5) 2014), pp. 1110-1122.
[40] James Leach, Jennifer P. Morton& Owen J. Sansom, “Neutrophils: Homing in on the myeloid mechanisms of metastasis,” Molecular Immunology, vol. 110 (2019), pp. 69-76.
[41] Lindsey J. Gay& Barbara Felding-Habermann, “Contribution of platelets to tumour metastasis,” Nature Reviews Cancer, vol. 11, no. 2 )2011), pp. 123-134.
[42]Joan Massague &Anna C. Obenauf, “Metastatic colonization by circulating tumour cells,” Nature, vol. 529, no. 7586 (2016), pp. 298-306.
[43] Fouad & Aanei, pp. 1016-1036.
[44]Nicolas Reymond, Bárbara Borda d'Agua& Anne J. Ridley, “Crossing the endothelial barrier during metastasis,” Nature Reviews Cancer, vol. 13, no. 12 (2013), pp. 858-870.
[45] Hamidi & Ivaska, pp.533 -548.
[46] Valastyan, Weinberg, pp. 275-292.
[47] Fouad & Aanei, pp. 1016-1036.
[48]Héctor Peinado, Simon Lavotshkin & David Lyden, “The secreted factors responsible for pre-metastatic niche formation: old sayings and new thoughts,” Seminars in cancer biology, vol. 21, no. 2 (2011).
[49]Bruno Costa-Silva et al., “Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver,” Nature Cell Biology, vol. 17, no. 6 (2015), pp. 816-826.
[50]Jae W. Lee et al., “Hepatocytes direct the formation of a pro-metastatic niche in the liver,” Nature, vol. 567, no. 7747) 2019), pp. 249-252.
[51]Elvin Wagenblast et al., “A model of breast cancer heterogeneity reveals vascular mimicry as a driver of metastasis,” Nature, vol. 520, no. 7547 (2015), pp3 .58-362.
[52] Roger R. Gomis & S. Gawrzak, “Tumor cell dormancy,” Molecular Oncology, vol. 11, no. 1 (2017), pp. 62-78.
[53]Filippo G. Giancotti, “Mechanisms governing metastatic dormancy and reactivation,” Cell, vol. 155, no. 4 (2013), pp. 750-7.64
[54] Ibid.
[55] Fouad & Aanei, pp. 1016-1036.
[56]Julio A. Aguirre-Ghiso & Maria Soledad Sosa, “Emerging topics on disseminated cancer cell dormancy and the paradigm of metastasis,” Annual Review of Cancer Biology, vol. 2 (2018), pp. 377-393.
[57]Maria Soledad Sosa, Emily Bernstein & Julio A. guirre-Ghiso, “Epigenetic regulation of cancer dormancy as a plasticity mechanism for metastasis initiation,” in: Yuzhuo Wang & Francesco Crea (eds.), Tumor Dormancy and Recurrence (Cham: Humana Press, 2017), pp. 1-16.
[58] Gomis, Roger R. & S. Gawrzak. “Tumor cell dormancy.” Molecular Oncology. vol. 11, no. 1 (2017). pp. 62-78.
[59] Yeh A. C. & Ramaswamy S., “Mechanisms of cancer cell dormancy—another hallmark of cancer?,” Cancer research, vol. 75, no. 23 (2015), pp. 50145122-.
[60] Aya Kobayashi et al., "Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone," Journal of Experimental Medicine, vol. 208, no. 13) 2011(, pp. 2641-2655.
[61] Ibid.
[62] Julio A. Aguirre-Ghiso et al., “Urokinase receptor and fibronectin regulate the ERK(MAPK) to p38(MAPK) activity ratios that determine carcinoma cell proliferation or dormancy in vivo,” Molecular Biology of the Cell, vol. 12, no. 4 (2001), pp. 863-879.
[63] Gomis & Gawrzak.
[64] Bragado P. et al., “TGF-β2 dictates disseminated tumour cell fate in target organs through TGF-β-RIII and p38α/β signalling,” Nature cell biology, vol. 15, no. 11 )2013), pp. 1351-1361.
[65] Cyrus M. Ghajar et al., “The perivascular niche regulates breast tumour dormancy,” Nature Cell Biology, vol. 15, no. 7 (2013), pp. 807-817.
[66] O. Straume et al., « Suppression of heat shock protein 27 induces long-term dormancy in human breast cancer,” Proceedings of the National Academy of Sciences of the United States of America, vol. 109, no. 22 (2012), pp. 8699-8704.
[67] Endo H. & Inoue M., “Dormancy in cancer,” Cancer Science, vol. 110, no. 2 (2019), pp. 474-480.
[68] Sosa, Bernstein & Aguirre-Ghiso, pp. 1-16.
[69]Giancotti, pp. 750-7.64
[70] Ibid.
[71]Jean Albrengues et al., “Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice,” Science, vol. 361, no. 6409 (2018).
[72]Dana Ishay-Ronen et al., “Gain Fat-Lose Metastasis: Converting Invasive Breast Cancer Cells into Adipocytes Inhibits Cancer Metastasis,” Cancer Cell, vol. 35, no. 1 (2019), pp. 17-32.
[73]Louise van der Weyden et al., “Genome-wide in vivo screen identifies novel host regulators of metastatic colonization,” Nature, vol. 541, no. 7636 (2017), pp. 233-336.
[74]Prudence Donovan et al., “Endovascular progenitors infiltrate melanomas and differentiate towards a variety of vascular beds promoting tumor metastasis,” Nature Communications, vol. 10, no .1 )2019), p. 18.
[75] Milan M. S. Obradovic et al., “Glucocorticoids promote breast cancer metastasis,” Nature, vol. 567, no. 7749 )2019), pp. 540-544.
[76]Ioanna Keklikoglou et al., “Chemotherapy elicits pro-metastatic extracellular vesicles in breast cancer models,” Nature Cell Biology, vol. 21, no. 2) 2019), pp. 190-202.
[77] Ibid.
[78] Ibid.
[79]Beth A. Helmink et al., “The microbiome, cancer, and cancer therapy,” Nature Medicine, vol. 25, no. 3 (2019), pp. 377-388.
[80] Smruti Pushalkar et al., “The Pancreatic Cancer Microbiome Promotes Oncogenesis by Induction of Innate and Adaptive Immune Suppression,” Cancer Discovery, vol. 8, no. 4) 2018), pp. 403-416.
[81] Concetta Panebianco, Angelo Andriulli & Valerio Pazienza, “Pharmacomicrobiomics: exploiting the drug-microbiota interactions in anticancer therapies,” Microbiome, vol. 6, no.1 )2018), p. 92.
[82] Jawad Fares, Mohamad Y. Fares & Youssef Fares, “Immune checkpoint inhibitors: Advances and impact in neuro-oncology,” Surgical Neurology International, vol. 10, no. 9 )2019 .(
[83] Albrengues et al.
[84]F. Stephen Hodi et al., “Improved survival with ipilimumab in patients with metastatic melanoma,” TheNew England Journal of Medicine, vol. 363, no. 8 (2010), pp. 711-723.
[85] Howard I. Scher et al., “Increased survival with enzalutamide in prostate cancer after chemotherapy,” TheNew England Journal of Medicine, vol. 367, no. 13) 2012), pp. 1187-1197 .
[86]Amye J. Tevaarwerk et al., “Survival in patients with metastatic recurrent breast cancer after adjuvant chemotherapy: little evidence of improvement over the past 30 years,” Cancer, vol. 119, no. 6 (2013), pp. 1140-1148.
[87] Albrengues et al.
[88]Jawad Fares et al., “Diagnostic Clinical Trials in Breast Cancer Brain Metastases: Barriers and Innovations,” Clinical Breast Cancer, vol. 19, no. 6 )2019), pp. 383-391.
[89]Imran Khan & Patricia S. Steeg, “Metastasis suppressors: functional pathways,” Laboratory Investigation, vol. 98, no. 2) 2018), pp. 198-210.
[90] Xiaohui Hu et al., “The RNA-binding protein AKAP8 suppresses tumor metastasis by antagonizing EMT-associated alternative splicing,” Nature Communications, vol. 11, no. 1 )2020), p. 486.
[91] Khan & Steeg.
[92] Ibid.
[93] Jean-Ckaude A. Marshall et al., “Effect of inhibition of the lysophosphatidic acid receptor 1 on metastasis and metastatic dormancy in breast cancer,” Journal of the National Cancer Institute, vol. 104 ,no. 17) 2012), pp. 1306-1319.
[94] Paul E. Goss & Ann F. Chambers, “Does tumour dormancy offer a therapeutic target?,” Nature Reviews Cancer, vol. 10, no. 12 (2010), pp. 871-877.
[95] Ilya Ulasov et al., “Editing cytoprotective autophagy in glioma: an unfulfilled potential for therapy,” Trends in Molecular Medicine, vol. 26, no. 3) 2020), pp. 252-262.
[96] Jawad Fares et al., “Current state of clinical trials in breast cancer brain metastases,” Neuro-Oncology Practice, vol. 6, no. 5 (2019), pp. 392-401.
[97] Jawad Fares et al., “Landscape of combination therapy trials in breast cancer brain metastasis,” InternationalJournal of Cancer, vol. 147, no. 7 (2020), pp. 1939-1952.
