الغلايكوجينين (Glycogenin) هو
إنزيم/
بروتين رئيس ضروري لتخليق جزيء الغلايكوجين (Glycogen) من جزيئات
الغلوكوز (Glucose)، وهو إنزيم مهم يبدأ عمليةَ
تخليق الغلايكوجين (Glycogenesis) من العدم، ومن دونه لا تستطيع الخلايا ربط جزيئات الغلوكوز معًا للبدء بالعملية. تعدّ عملية تخليق الغلايكوجين أمرًا حيويًا للحفاظ على توازن الطاقة، والاستجابة لمتطلبات
التمثيل الغذائي في الجسم. ويُعَد بروتين الغلايكوجينين إنزيمًا فريدًا، إذ إنه بعد تحفيز التفاعل البيوكيميائي، يظل مرتبطًا بالمنتوج ولا يتركه. لقد أدى اكتشاف هذا البروتين الفريد وتوصيفه إلى تطوير فهم
استقلاب الغلايكوجين وآثاره في الصحة والمرض بشكل كبير، فنقصانه يعدّ أحد
اضطرابات تخزين الغلايكوجين الكثيرة، ما يسلط الضوء على أهميته في فيزيولوجيا
الخلية وصحة الإنسان. ولإنزيم الغلايكوجينين اسم آخر متعارف عليه، هو غلايكوجينين غلوكوزيل ترانسفيراز (Glycogenin transerase glucosyl)، أو الاسم العلمي: (UDP-α-D-glucose: Glycogenin α-D-glucosyltransferase).
اكتشافه
يمكن إرجاع اكتشاف الغلايكوجينين إلى الأبحاث المبكّرة، في المدة ما بين منتصف القرن التاسع عشر - باكتشاف كلود برنارد (1813-1878) جزيء
الغلايكوجين[1] - وأوائل القرن العشرين عندما بدأ الباحثون في استنتاج بنية الغلايكوجين واستقلابه[2]. والغلايكوجين
سكر متعدّد (Polysaccharide) متفرع ومعقّد، مكوّن من جزيئات غلوكوز متصلة بعضها ببعض، ويعمل بوصفه شكلًا من أشكال تخزين الغلوكوز في الحيوانات، بما في ذلك البشر. في عام 1939، اكتشف كارل إف كوري (1896-1984) وغيرتي كوري (1896-1957) إنزيم غلايكوجين فوسفوريلاز (Glycogen phosphorylase)، الذي يُحلِّل الغلايكوجين إلى جزيئات الغلوكوز 1- فوسفات[3]. وفي عام 1957، اكتشف لويس ليلوار (1906-1987) وكارلوس كارديني إنزيم تصنيع الغلايكوجين: غلايكوجين سينثاز (Glycogen synthase)[4]. وعملت كلارا كريسمان على استقلاب الغلايكوجين باستخدام ركائز الغلوكوز المشعّة في سبعينيات القرن العشرين، وأوضحت الحاجة إلى وجود بروتين أوّلي مهمته البدء في تخليق الغلايكوجين[5].
في عملها في المجال، وجدت كريسمان ورينيه بارينغو أن ثمة ثلاثة إنزيمات منفصلة تبدأ بتخليق الغلايكوجين[6]. ولاحقًا، لحِظ ويليام ويلان (1924-2021) وزملاؤه وجود بروتين داخل جزيء الغلايكوجين، وذلك في أثناء محاولة استخلاص الغلايكوجين وتنقيته من
العضلات الهيكلية للأرنب، في ظل ظروف بيوكيميائية تُزيل جميع البروتينات غير المرتبطة تساهميًا. ثم أظهر ميغيل أون وخوان كرتينو أن الغلايكوجين المرتبط بالبروتين يرتبط بالحمض الأميني التيروسين (Tyrosine)[7]. وقد سمحت التطورات اللاحقة في التقنيات
الكيميائية الحيوية والبيولوجيا الجزيئية بعزل هذا البروتين وتوصيفه وتسميته الغلايكوجينين، بوساطة ويلان عام 1985[8].
أسهمت الاكتشافات الرئيسة في فهم تخليق الغلايكوجين، مثل تحديد الإنزيمات المشاركة في
المسار الأيضي بيوكيميائيًا وجزيئيًا، إسهامًا كبيرًا في كشف دور الغلايكوجينين، وقدّم الباحثون الأساسيون في هذا المجال، بمن فيهم ويلان وفريتز ليبمان (1899-1986) وآخرون، إسهامات كبيرة في توضيح الآليات البيوكيميائية لاستقلاب الغلايكوجين[9]، وتسليط الضوء على أهمية هذا البروتين في هذه العملية.
شكله وهيكله
الغلايكوجينين بروتين
ثنائي متماثل {{البروتين ثنائي التماثل: (Homodimer) معقّد بروتيني يتكون من وحدتين فرعيتين متطابقتين (مونومرات) ترتبطان ببعضهما، غالبًا من خلال تفاعلات غير تساهمية، أو عبر الروابط ثنائية الكبريتيد (Disulfide bonds) التساهمية. تعمل هذه الوحدات الفرعية المزدوجة بشكل تعاوني، تشيع ثنائيات التماثل في الإنزيمات، وعوامل النسخ والمستقبِلات، وتؤدي أدوارًا في الإشارات الخلوية والمسارات الأيضية.}} يتكوّن من وحدتين فرعيتين متشابهتين، كل واحدة وزنها الجزيئي 37
كيلو دالتون {{الدالتون: (Da) وحدة كتلة ذرية، تساوي واحدًا على اثني عشر من كتلة ذرة الكربون-12، وتُستخدم للتعبير عن الأوزان الجزيئية والذرية. أما الكيلو دالتون (kDa)، فهو وحدة كتلة جزيئية تساوي 1000 دالتون، ويُستخدم عادةً للتعبير عن حجم البروتينات والأحماض النووية، والجزيئات الحيوية الكبيرة الأخرى.}}.
للغلايكوجينين بنيةٌ هيكلية تتضمن
مجالات بروتينية وظيفية {{المجالات البروتينية الوظيفية: (Functional domains) هي مناطق مستقلة بنيويًا داخل البروتين، تؤدي مهمات محددة، مثل ربط الجزيئات، أو تحفيز التفاعلات، أو التوسط في الارتباطات. تحدد هذه الوحدات الدور البيولوجي للبروتين، ويمكنها أن تتطور بشكل مستقل.}} ، وأنماطًا بنيوية {{الأنماط البنيوية: (Structural Motifs)
هي ترتيبات متكررة ومستقرة في هياكل البروتين الثانوية (مثل: لولب ألفا وصفائح بيتا المتثنية)، تشكل وحدات وظيفية أو مُثَبِّتة. ومن الأمثلة على ذلك: لولب-لفّة-لولب (Helix-turn-helix)، ودبوس الشعر بيتا (Beta hairpin)، وإصبع الزنك (Zinc finger)، التي غالبًا ما تكون لها أدوار للارتباط بالحمض النووي أو التحفيز، أو تفاعلات البروتين مع البروتين.}} أساسية لوظيفتها في تخليق الغلايكوجين[10]. حُلَّت بنية الغلايكوجينين باستخدام شكلين بلوريين مختلفين[11]، ويعمل البروتين بوصفه بادِئًا (Primer) لتكوين الغلايكوجين، إذ يبدأ بربط وحدات الغلوكوز على نفسها قبل استطالة سلسلة الغلايكوجين. يحتوي
المجال التحفيزي {{المجال التحفيزي: (Catalytic domain) هو منطقة الإنزيم أو البروتين المسؤولة عن نشاطه البيوكيميائي، وتحتوي على الموقع النشط الذي ترتبط عليه الركيزة وتحدث فيه عملية التحفيز. وغالبًا ما تكون له بنية محفوظة تطوريًا، وهي ضرورية لوظيفته.}} للغلايكوجينين على
الموقع النشط {{الموقع النشط: (Active site) هو منطقة محددة في الإنزيم، حيث ترتبط جزيئات الركيزة وتخضع لتفاعل كيميائي، غالبًا من خلال ترتيبات دقيقة للأحماض الأمينية التي تحفز التفاعل الكيميائي.}} المسؤول عن نقل جزيء الغلوكوز من مركب غلوكوز ثنائي فوسفات اليوريدين (UDP-glucose) إلى نفسه، مكوِّنًا بادئ الغلايكوجين، ويعدّ هذا البادئ نقطة البداية لاستطالة الغلايكوجين اللاحقة بوساطة إنزيم غلايكوجين سينثاز.
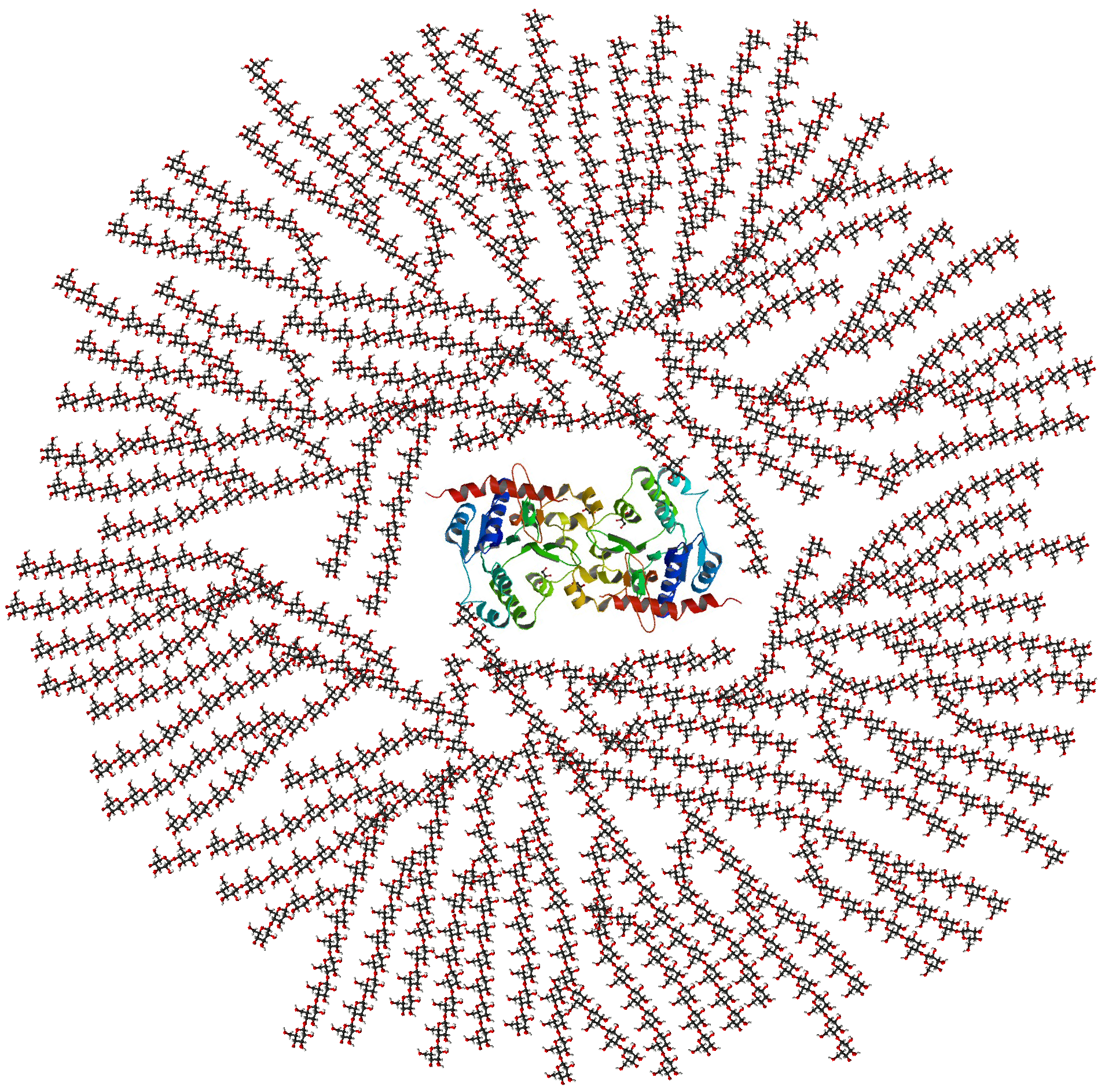 بروتين غلايكوجينين ثنائي متماثل (في الوسط) يصنّع جزيء الغلايكوجين (المحيط) ويظل مرتبطًا به
بروتين غلايكوجينين ثنائي متماثل (في الوسط) يصنّع جزيء الغلايكوجين (المحيط) ويظل مرتبطًا به
حذف الصورة؟
سيؤدي هذا إلى نقل الصورة إلى سلة المهملات.
في
الأنسجة العضلية، يتضمن بدء تخليق الغلايكوجين إنزيمين رئيسين: غلايكوجينين -1، المسؤول عن ابتداء جزيء الغلايكوجين، من خلال
الغلكزة الذاتية {{الغلكزة الذاتية: (Autoglucosylation) عملية إنزيمية تؤدي إلى ربط جزيئات كربوهيدراتية على الإنزيم نفسه (يغلكز الإنزيم ذاته)، ما يؤدي غالبًا إلى تعديل وظيفتها أو استقرارها أو تفاعلاتها.}}؛ وغلايكوجين سينثاز -1، الذي يُكَبِّر سلسلة الغلايكوجين. يرتبط الإنزيمان في وحدة وظيفية معًا لأداء وظيفة تخليق الغلايكوجين. وقد أتاحت التطورات الحديثة في
المجهر الإلكتروني فائق البرودة {{المجهر الإلكتروني فائق البرودة: (Cryo-Electron microscopy) تقنية تصوير تكشف عن الهياكل ثلاثية الأبعاد للجزيئات الحيوية بدقة شبه ذريّة، إذ تُجمَّد العينات بسرعة للحفاظ على حالتها الأصلية، ثم تُصوّر باستخدام مجهر إلكتروني. وقد ابتكر هذه التقنية دوبوشيه، وفرانك، وهندرسون، فمنحوا في إثر ابتكارهم جائزة نوبل في الكيمياء. تتجاوز هذه التقنية الحاجة إلى التبلور، فتتيح دراسة المعقدات البروتينية الكبيرة، والڨيروسات، والآليات الخلوية الأخرى.}} إجراء فحوصات تفصيلية لمعقّد غلايكوجين سينثاز-1-غلايكوجينين في الخلايا البشرية. في المجهر الإلكتروني، وجد الباحثون عدة أشكال للمعقّد، ما كشف أن شكل الغلايكوجين سينثاز هو رباعي (Tetramer) مرن قادر على تعديل شكله، ومن ثم، هناك إمكانية للوصول إلى مواقعه النشطة استجابةً للتحفيز. في شكل محدد من هذه الأشكال المختلفة، تترابط
المجالات التحفيزية للغلايكوجينين-1 والغلايكوجين سينثاز-1، ما يشير إلى أن هذا الترتيب يُقرّب المواقع النشطة من بعضها، لتسهيل إجراء التفاعلات المتتالية المرتبطة. إن هذا الشكل البنائي للمعقّد، غالبًا ما يمكّن الغلايكوجين سينثاز-1 من استقبال بادئ الغلايكوجين المربوط على الغلايكوجينين-1، ما يقدم نموذجًا لكيفية البدء في المراحل الرئيسة لتخليق الغلايكوجين، بدءًا من تكوين البادئ وحتى استطالة السلسلة[12].
تصنيف الإنزيم ونظرائه والتوزيع في الأنسجة
 التفاعل الكيميائي للغلايكوجينين
التفاعل الكيميائي للغلايكوجينين
حذف الصورة؟
سيؤدي هذا إلى نقل الصورة إلى سلة المهملات.
يُصنَّف الغلايكوجينين تحت
رقم التصنيف الإنزيمي {{
رقم التصنيف الإنزيمي:
(Enzyme commission number)، (رقم لجنة الإنزيم/ EC number) وهو نظام تصنيف رقمي للإنزيمات، يعتمد على تفاعلاتها التحفيزية، ويحتوي على أربعة أرقام تُحدّد نوع التفاعل والركيزة، وأمورًا تفصيلية أخرى.}}EC 2.4.1.186[13]، فهو يتبع عائلة الترانسفيراز (Transferase) الناقلة للمجموعات الوظيفية. ويعمل هذا الإنزيم بوصفه ناقلًا لمجموعة غلوكوز، فهو يؤدي عملية
غلكزة ذاتية. لهذا البروتين نظيران: الغلايكوجينين-1 (المشفَّر بوساطة
جين GYG1)، وهو موجود أساسًا في
الأنسجة العضلية؛ والغلايكوجينين-2 (الذي يشفّره جين GYG2)، وهو السائد في
الكبد وعضلة القلب[14]. يعكس هذا التوزيع الخاص بالأنسجة الأدوار الأيضية المتنوعة للغلايكوجينين في سياقات فيزيولوجية مختلفة.
يحفّز الغلايكوجينين التفاعل الكيميائي الذي يتضمن
الغلكزة الذاتية للبروتين نفسه، على النحو الذي ينقل فيه جزيء الغلوكوز من جزيء الغلوكوز ثنائي فوسفات اليوريدين إلى نفسه، ويرتبط به برابطة تساهمية عبر الحمض الأميني تيروسين رقم 194 من سلسلة الأحماض الأمينية في الغلايكوجينين. ومن ثم، يستخدم غلوكوزًا آخر ثنائي فوسفات اليوريدين لإضافته إلى الغلوكوز الأول، ويستمر في الإضافة حتى يصل طول السكر إلى 7 أو 8 وحدات غلوكوز متتالية، ما يؤدي إلى تخليق
مبلمر غلوكوز قصير، مرتبط بالتيروسين داخل بنية بروتين الغلايكوجينين. إن مبلمر الغلوكوز القصير، المرتبط ببروتين الغلايكوجينين، هو البادئ الذي يمكن لإنزيم الغلايكوجين سينثاز العمل عليه لتكوين الغلايكوجين.
يمكن تلخيص التفاعل الكيميائي على النحو التالي:
UDP-glucose + Glycogenin(Tyr194)-OH → Glycogenin(Tyr194)-O-(glucose) + UDP
وظيفة الغلايكوجينين وأهميته السريرية
يعدّ النشاط الإنزيمي للغلايكوجينين فريدًا من نوعه، لأنه يضيف وحدة البناء الأساسية (الغلوكوز) إلى نفسه، ويستمر في بناء مزيد من وحدات الغلوكوز عليه، قبل تسليم عملية تخليق الغلايكوجين إلى إنزيم غلايكوجين سينثاز. ويظل الغلايكوجينين مدفونًا داخل جزيء الغلايكوجين المتنامي، لذا، يؤدي الغلايكوجينين أدوارًا أساسية في بدء تخليق الغلايكوجين بتحفيز تكوين سلسلة قصيرة من السكريات القليلة، التي تعمل بمنزلة البادئ لاستطالة الغلايكوجين. وتعدّ هذه السلسلة البادئة ضرورية لإنزيم الغلايكوجين سينثاز، لإضافة وحدات الغلوكوز واستطالة الجزيء. ويذكر أن ثمة إنزيمًا آخر في تخليق الغلايكوجين، يُسمّى
إنزيم تفريع الغلايكوجين {{إنزيم تفريع الغلايكوجين (EC 2.4.1.18): (Glycogen branching enzyme) إنزيم ناقل لمجموعة غلوكوز، يُدخل نقاط تفرّع في جزيء الغلايكوجين في أثناء تصنيعه، ويُكوّن روابط غلوكوسيدية بين كربون 1 من جزيء غلوكوز، وذرة كربون 6 من جزيء غلوكوز آخر (روابط ألفا 1ß6)، ما يُعزز قابلية الذوبان والوصول الإنزيمي لتكسير الغلوكوز بسرعة. يُسبب نقص هذا الإنزيم مرض تخزين الغلايكوجين من النوع الرابع.}}، الذي يضيف تفرعات لجزيء الغلايكوجين، ما يؤدي إلى تكوين البنية المتفرعة المميزة لسلاسل الغلايكوجين. تُنظَّم التفاعلات بين الإنزيمات الثلاثة في تخليق الغلايكوجين، ولا سيما بين الغلايكوجين سينثاز والغلايكوجينين، بإحكام، ما يضمن إنتاج الغلايكوجين وتخزينه بشكل فعّال، استجابة لاحتياجات الطاقة في الخلية. علاوة على ذلك، يمتد دور الغلايكوجينين إلى ما هو أبعد من تخليق الغلايكوجين، فهو يشارك في تنظيم استقلاب الغلايكوجين، ما يسهم في التوازن بين تخليق الغلايكوجين وتكسيره {{تحلل الغلايكوجين: (Glycogenolysis) هو المسار الأيضي الذي يُكسِّر جزيء الغلايكوجين إلى جزيئات أصغر، وإلى جزيئات غلوكوز1- فوسفات وغلوكوز حر، باستخدام إنزيم غلايكوجين فوسفوريلاز، وإنزيم إزالة تفريع الغلايكوجين، ما يُوفر طاقة سريعة في أثناء الصيام أو التمرين. يُنظَّم هرمونيًا بوساطة هرمونَي الغلوكاجون والأدرينالين.}} لاستخلاص الطاقة.
يمكن أن تؤدي المشكلات في وظيفة الغلايكوجينين أو تنظيم عمله، إلى أمراض وراثية مثل
اضطرابات تخزين الغلايكوجين {{اضطرابات تخزين الغلايكوجين: (Glycogen storage disorders) مجموعة من الحالات النادرة التي لا يستطيع فيها الجسم استخدام الغلايكوجين أو تخزينه بشكل صحيح، وهي أنواع من اضطرابات التمثيل الغذائي الوراثية.}} (الجدول 1)[15]، إذ لا يتمكن الأفراد المصابون من تخزين الغلايكوجين و/ أو استعماله بشكل صحيح. ومرض تخزين الغلايكوجين، النوع 15، يرتبط مباشرةً بنقص الغلايكوجينين، ما يُسلّط الضوء على أهمية هذا البروتين في الحفاظ على توازن الغلايكوجين والصحة الأيضية العامة للخلية وللكائن[16]. في هذا المرض المسمّى أيضًا مرض نقص الغلايكوجينين (خلل في جين GYG1)، حُدِّدت بعض
الطفرات التي تؤثر في وظيفة الغلايكوجينين، ولا سيما في
الأنسجة العضلية لدى المرضى الذين يعانون منه، ما يؤدي إلى أعراض مثل ضعف
العضلات وأمراض القلب، بسبب ضعف استقلاب الغلايكوجين. وتتشابه أعراض الطفرات التي تؤدي إلى نقص الغلايكوجينين مع نقص الغلايكوجين سينثاز في العضلات، بما في ذلك عدم تحمل التمارين الرياضية، وضعف العضلات وفقدان كتلها، وفي الحالات الشديدة قد تؤدي إلى فشل القلب[17].
[الجدول 1] - اضطرابات تخزين الغلايكوجين
نوع المرض
|
الإنزيم الناقص |
الاسم (بحسب المكتشِف) |
أجهزة الجسم الرئيسة المتضررة |
|---|
نوع صفر أ | غلايكوجين سينثاز 2 (Glycogen synthase 2) | داء لويس (Lewis’ disease) | الكبد |
نوع صفر ب | غلايكوجين سينثاز العضلي (Muscle glycogen synthase) | داء لويس (Lewis’ disease) | العضلات الهيكلية والقلبية |
نوع 1أ | غلوكوز 6- فوسفاتاز (Glucose 6-phosphatase) | داء فون غيركه (von Gierke disease) | الكبد |
نوع 1ب | ناقل الغلوكوز 6- فوسفاتاز (Glucose 6-phosphatase transporter) | داء فون غيركه (von Gierke disease) | الكبد |
نوع 2أ
| المالتاز الليسوسومي الحمضي (Lysosomal acid maltase) | داء بومبه (Pompe dissease) | العضلات الهيكلية والقلبية |
نوع 2ب | لامب-2 (Lysosome-associated membrane protein 2) | داء بومبه (Pompe disease) | العضلات الهيكلية والقلبية |
نوع 3 | إنزيم إزالة تفريع الغلايكوجين (Glycogen debranching enzyme) | داء فوربس-كوري (Forbes-Cori disease) | الكبد |
نوع 4 | إنزيم تفريع الغلايكوجين (Glycogen branching enzyme) | داء أندرسن (Andersen’s disease) | الكبد |
نوع 5 | الفوسفوريلاز العضلي (Muscle phosphorylase) | داء مكاردل (McArdle’s disease) | العضلات الهيكلية |
نوع 6 | الفوسفوريلاز الكبدي (Liver phosphorylase) | داء هيرز (Hers’ disease) | الكبد |
نوع 7 | فوسفوفركتو كيناز (Phosphofructokinase) | داء تاروي (Tarui disease) | العضلات الهيكلية |
نوع 8 | غلايكوجين فوسفوريلاز-ب-كيناز الكبدي (Liver glycogen phosphorylase-b-kinase) | | الكبد |
نوع 9أ | فوسفوريلاز كيناز (Phosphorylase kinase) | | الكبد |
نوع 10 | فوسفوغليسيرات ميوتاز (Phosphoglycerate mutase) | | العضلات الهيكلية |
نوع 11 | ناقل الغلوكوز 2 (GLUT2) | متلازمة فانكوني-بيكل (Fanconi-Bickel syndrome) | الكبد |
نوع 12 | ألدولاز أ (Aldolase A) | | العضلات الهيكلية |
نوع 13 | بيتا-إينولاز (Beta-enolase) | | العضلات الهيكلية |
نوع 14 | فوسفوغلوكو ميوتاز-1 (Phosphoglucomutase-1)
| | العضلات الهيكلية |
نوع 15 | غلايكوجينين-1 (Glycogenin-1) | | العضلات الهيكلية والقلبية |
الأبحاث والتطبيقات
تستمر الأبحاث الجارية بشأن الغلايكوجينين والغلايكوجين وأمراض تخزينهما المختلفة في الكشف عن معلومات جديدة عن وظائف هذه الجزيئات وتنظيمها، وتطبيقاتها العلاجية المحتملة. توفر الدراسات التي تبحث في دور الغلايكوجينين في
أمراض الغلايكوجين الأيضية، بوصفه هدفًا مباشرًا أو غير مباشر لتطوير الأدوية، مسارًا واعدًا للبحث المستقبلي والتدخلات السريرية للمرضى. وتشمل التطبيقات الحديثة الحالية للبحث في هذا المجال مجموعةً من التقنيات والأساليب التي تهدف إلى فهم الآليات الجزيئية الكامنة وراء هذه الحالات، وتطوير التدخلات العلاجية لها وتوفيرها. تشمل بعض المجالات الرئيسة الأبحاث الجارية والتطبيقات ما يلي:
- 1.
البيولوجيا الهيكلية {{البيولوجيا الهيكلية (علم الأحياء البنيوي): (Structural biology) دراسة البنية ثلاثية الأبعاد للجزيئات الحيوية الكبيرة (مثل البروتينات والأحماض النووية والمعقَّدات)، باستخدام تقنيات مثل: دراسة البلورات بالأشعة السينية، والمجهر الإلكتروني فائق البرودة، والرنين المغناطيسي النووي. يكشف هذا العلم كيف تحدد البنى الجزيئية وظيفة الجزيئات، ما يُساعد في تصميم الأدوية وفهم آلية الأمراض.}} والنمذجة الجزيئية {{النمذجة الجزيئية: (Molecular modeling) وسيلة حاسوبية لمحاكاة وتصوير البنية ثلاثية الأبعاد للجزيئات وديناميكياتها وتفاعلاتها. وتستخدم تقنيات مثل محاكاة الديناميكيات الجزيئية (Molecular dynamics simulations) وميكانيكا الكم، للتنبؤ بسلوك الجزيئات، وتحسين تصميم الأدوية، واستكشاف الآليات البيوكيميائية.}}: تُستخدَم التقنيات المتقدمة في البيولوجيا الهيكلية، مثل
المجهر الإلكتروني فائق البرودة ودراسة البلورات بالأشعة السينية {{علم دراسة البلورات بالأشعة السينية: (X-ray crystallography) تقنية مخبرية لتحديد البنية الذرية لبلورات المواد البيولوجية، عن طريق تحليل أنماط حَيْد الأشعة السينية عن البلورات البيولوجية (البروتينات أو الأحماض النووية). ويكشف هذا عن الترتيب ثلاثي الأبعاد للجزيئات، ما يُمكّن من تحقيق إنجازات في تصميم الأدوية وفهم الآليات البيولوجية. كانت روزاليند فرانكلين رائدة هذه التقنية في دراستها لِلّولب الحمض النووي (DNA).}}، لدراسة الهياكل ثلاثية الأبعاد للغلايكوجينين، وارتباطاته مع الإنزيمات الأخرى المشاركة في تخليق الغلايكوجين وتفاعلاته معها[18]. كذلك تساعد تقنيات
النمذجة الجزيئية في التنبؤ بالتغيرات الهيكلية لهذا البروتين في مختلف الحالات، علاوة على محاولة فهم الأهمية الوظيفية للطفرات المرتبطة بأمراض تخزين الغلايكوجين.
- 2.
علم الجينوم (Genomics) والتحليل الوراثي (Genetic analysis): تُستخدَم تقنيات
التسلسل عالي الإنتاجية {{التسلسل عالي الإنتاجية
(HTS): (High-throughput sequencing) يشير هذا المصطلح إلى تقنيات متقدمة لفحص تسلسل الحمض النووي الريبي منقوص الأكسجين/ الحمض النووي الريبي (DNA/RNA) (مثل Illumina وPacBio)، تُولِّد كميات هائلة من البيانات الجينية بسرعة وبتكاليف منخفضة. تُستخدَم هذه التقنيات في علم الجينوم، وعلم النسخ، وعلم الميتاجينوم، في تطبيقات مثل الكشف عن المتحوّرات وتحليل التعبير الجيني ومراقبة مسببات الأمراض.}}، بما في ذلك
تسلسل الإكسوم الكامل {{تسلسل الإكسوم الكامل (WES): (Whole-exome sequencing) تقنية جينومية تُحلِّل مناطق ترميز البروتين (الإكسونات/ exons) في الجينات جميعها (ما يقارب 1-2 في المئة من الجينوم)، لتحديد الطفرات المسببة للأمراض. تُوازِن هذه التقنية بين التكلفة والعمق، ما يُساعد في تشخيص الاضطرابات الوراثية النادرة والسرطان والأمراض المعقدة. كذلك فإنها تكشف عن المتغيرات ذات الصلة سريريًا بشكل أسرع من تسلسل الجينوم الكامل.}}، ودراسة الترابط الجينومي الكامل (Genome-wide association studies)، لتحديد المتغيرات والطفرات الجينية المرتبطة بأمراض تخزين الغلايكوجين[19]. وتعدّ هذه الأبحاث ضروريةً لدراسة آليات المرض ومسبباته ولتطوير استراتيجيات علاجية شخصية.
- 3. النماذج الخلوية والحيوانية (Cellular and animal models): يستخدم الباحثون نماذج
زراعة الخلايا، مثل
الخلايا الأولية {{الخلايا الأولية: (Primary cells) خلايا تُعزَل مباشرة من أنسجة حية وتُزرَع في المختبر. وعلى عكس سلالات الخلايا المُخلَّدة (Immortalized cell lines)، تحتفظ هذه الخلايا بخصائصها الأصلية، لكن عدد انقساماتها محدود. وتُستخدَم على نطاق واسع في أبحاث اختبارات الأدوية، وعلم أحياء السرطان، بوصفها نماذج ذات صلة فيزيولوجية، وهي أكثر دقة بيولوجيًا من سلالات الخلايا المُخلَّدة، ولكن يصعب الحفاظ عليها.}}، والخلايا المستمدّة من المرضى الذين يعانون من أمراض تخزين الغلايكوجين المختلفة، لدراسة مسببات المرض، ومساراته الجزيئية، والأهداف الخلوية العلاجية المحتمَلة. تُستخدم أيضًا النماذج الحيوانية، بما في ذلك الفئران المعدّلة وراثيًا والكائنات الحية الأخرى، لدراسة التأثيرات الفيزيولوجية لاختلال الغلايكوجينين، واختبار التدخلات العلاجية الجديدة. إلى جانب ذلك، ثمة نماذج حيوانية لنقص الغلايكوجينين تساعد في دراسة هذه الأمراض الوراثية[20].
- 4. الأساليب العلاجية: تُدرَس استراتيجيات علاجية جديدة لاضطرابات تخزين الغلايكوجين، بما في ذلك
العلاج الجيني (Gene therapy)، والعلاج الإنزيمي التعويضي {{العلاج الإنزيمي التعويضي
(ERT): (Enzyme replacement therapy) علاج للاضطرابات الأيضية الوراثية، مثل أمراض التخزين في الأجسام الحالة (Iysosomal storage disorders)، إذ يُستكمَل الإنزيم الناقص أو غير الفعّال عن طريق الحقن الوريدي بإنزيمات وظيفية مُنتَجة مخبريًا. يُخفف هذا العلاج الأعراض باستعادة الوظيفة الأيضية، مع أنه غالبًا ما يتطلب العلاج مدى الحياة، وهو باهظ التكلفة.}}، والمثبطات أو المنشطات التي تستهدف إنزيمات استقلاب الغلايكوجين، علاوة على التقنيات الحديثة في التحرير الجيني (Gene editing)، مثل
كريسبر/كاس9 (CRISPR-Cas9)[21]. ولتقييم سلامة هذه التدخلات العلاجية وفاعليتها، تُجرى كثير من الدراسات قبل السريرية والتجارب السريرية.
المراجع
Almodóvar-Payá, Aitana et al. “Preclinical Research in Glycogen Storage Diseases: A Comprehensive Review of Current Animal Models.”
International Journal of Molecular Sciences. vol. 21, no. 24, article no. 9621 (2020).
Aon, Miguel A. & Juan A. Curtino. “Protein-Bound Glycogen Is Linked to Tyrosine Residues.”
Biochemical Journal. vol. 229, no. 1 (1985). pp. 269-272.
Cardini, C. E. & Luis F. Leloir. “Enzymatic Formation of Acetylgalactosamine.”
Journal of Biological Chemistry. vol. 225, no. 1 (1957). pp. 317-324. at:
https://acr.ps/1L9BPuf
“Glycogen Storage Disease (GSD).” Cleveland Clinic. at:
https://acr.ps/1L9BP95
Cori, Carl F., Gerhard Schmidt & Gerty T. Cori. “The Synthesis of a Polysaccharide from Glucose-1-Phosphate in Muscle Extract.”
Science. vol. 89, no. 2316 (1939). pp. 464-465.
Fastman, Nathan M. et al. “The Structural Mechanism of Human Glycogen Synthesis by the GYS1-GYG1 Complex.”
Cell Reports. vol. 40, no. 1, article no. 111041 (2022).
Gardiner, A. et al. “Glycogen Storage Disease Type XV: A Case Report.”
Neuromuscular Disorders. vol. 25 (2015).
Gibbons, Brian J., Peter J. Roach & Thomas D. Hurley. “Crystal Structure of the Autocatalytic Initiator of Glycogen Biosynthesis, Glycogenin.”
Journal of Molecular Biology. vol. 319, no. 2 (2002). pp. 463-477.
Koeberl, Dwight D. et al. “Gene Therapy for Glycogen Storage Diseases.”
Journal of Inherited Metabolic Disease. vol. 47, no. 1 (2024). pp. 93-118.
Krisman, Clara R. “A Possible Intermediate in the Initiation of Glycogen Biosynthesis.”
Biochemical and Biophysical Research Communications. vol. 46, no. 3 (1972). pp. 1206-1212.
Krisman, Clara R. & Renée Barengo. “A Precursor of Glycogen Biosynthesis: Alpha-1,4-Glucan-Protein.”
European Journal of Biochemistry. vol. 52, no. 1 (1975). pp. 117-123.
Litwack, Gerald.
Human Biochemistry. 2nd ed. Boston: Academic Press, 2022.
Mancheño, Nuria et al. “Cardiac Phenotype in Glycogen Storage Disease Type XV: A Rare Cardiomyopathy to Bear in Mind.”
Revista Española de Cardiología (English Edition). vol. 74, no. 1 (2021). pp. 99-101.
Marr, Laura et al. “Mechanism of Glycogen Synthase Inactivation and Interaction with Glycogenin.”
Nature Communications. vol. 13, no. 1, article no. 3372 (2022).
“PubChem Enzyme Summary for Enzyme 2.4.1.186, Glycogenin Glucosyltransferase (EC 2.4.1.186).” National Center for Biotechnology Information. at:
https://acr.ps/1L9BPnQ
Robbins, Phillips W., Robert R. Traut & Fritz Lipmann. “Glycogen Synthesis from Glucose, Glucose-6-Phosphate, and Uridine Diphosphate Glucose in Muscle Preparations.”
Proceedings of the National Academy of Sciences. vol. 45, no. 1 (1959). pp. 6-12.
Rodriguez, Ignacio R. & William J. Whelan. “A Novel Glycosyl-Amino Acid Linkage: Rabbit-Muscle Glycogen Is Covalently Linked to a Protein via Tyrosine.”
Biochemical and Biophysical Research Communications. vol. 132, no. 2 (1985). pp. 829-836.
Smythe, Carl & Philip Cohen. “The Discovery of Glycogenin and the Priming Mechanism for Glycogen Biogenesis.”
European Journal of Biochemistry. vol. 200, no. 3 (1991). pp. 625-631.
Visuttijai, Kittichate et al., “Glycogenin Is Dispensable for Glycogen Synthesis in Human Muscle, and Glycogenin Deficiency Causes Polyglucosan Storage.”
Journal of Clinical Endocrinology & Metabolism. vol. 105, no. 2 (2020). pp. 557-566.
Ying, Shen et al., “Molecular Diagnosis of Panel-Based Next-Generation Sequencing Approach and Clinical Symptoms in Patients With Glycogen Storage Disease: A Single Center Retrospective Study.”
Frontiers in Pediatrics. vol. 8, article no. 600446 (2020).
Young, F. G. “Claude Bernard and the Discovery of Glycogen.”
British Medical Journal. vol. 1, no. 5033 (1957). pp. 1431-1437.
[1] F. G. Young, “Claude Bernard and the Discovery of Glycogen,”
British Medical Journal, vol. 1, no. 5033 (1957), pp. 1431-1437.
[2] Carl Smythe & Philip Cohen, “The Discovery of Glycogenin and the Priming Mechanism for Glycogen Biogenesis,”
European Journal of Biochemistry, vol. 200, no. 3 (1991), pp. 625-631.
[3] Carl F. Cori, Gerhard Schmidt & Gerty T. Cori, “The Synthesis of a Polysaccharide from Glucose-1-Phosphate in Muscle Extract,”
Science, vol. 89, no. 2316 (1939), pp. 464-465.
[4] C. E. Cardini & Luis F. Leloir, “Enzymatic Formation of Acetylgalactosamine,”
Journal of Biological Chemistry, vol. 225, no. 1 (1957), pp. 317-324, at:
https://acr.ps/1L9BPuf
[5] Clara R. Krisman, “A Possible Intermediate in the Initiation of Glycogen Biosynthesis,”
Biochemical and Biophysical Research Communications, vol. 46, no. 3 (1972), pp. 1206-1212.
[6] Clara R. Krisman & Renée Barengo, “A Precursor of Glycogen Biosynthesis: Alpha-1,4-Glucan-Protein,”
European Journal of Biochemistry, vol. 52, no. 1 (1975), pp. 117-123.
[7] Miguel A. Aon & Juan A. Curtino, “Protein-Bound Glycogen Is Linked to Tyrosine Residues,”
Biochemical Journal, vol. 229, no. 1 (1985), pp. 269-272.
[8] Ignacio R. Rodriguez & William J. Whelan, “A Novel Glycosyl-Amino Acid Linkage: Rabbit-Muscle Glycogen Is Covalently Linked to a Protein via Tyrosine,”
Biochemical and Biophysical Research Communications, vol. 132, no. 2 (1985), pp. 829-836.
[9] Phillips W. Robbins, Robert R. Traut & Fritz Lipmann, “Glycogen Synthesis from Glucose, Glucose-6-Phosphate, and Uridine Diphosphate Glucose in Muscle Preparations,”
Proceedings of the National Academy of Sciences, vol. 45, no. 1 (1959), pp. 6-12.
[10] Gerald Litwack,
Human Biochemistry, 2nd ed. (Boston: Academic Press, 2022), pp. 183-205.
[11] Brian J. Gibbons, Peter J. Roach & Thomas D. Hurley, “Crystal Structure of the Autocatalytic Initiator of Glycogen Biosynthesis, Glycogenin,”
Journal of Molecular Biology, vol. 319, no. 2 (2002), pp. 463-477.
[12] Nathan M. Fastman et al., “The Structural Mechanism of Human Glycogen Synthesis by the GYS1-GYG1 Complex,”
Cell Reports, vol. 40, no. 1, article no. 111041 (2022).
[13] “PubChem Enzyme Summary for Enzyme 2.4.1.186, Glycogenin Glucosyltransferase (EC 2.4.1.186),” National Center for Biotechnology Information, accessed on 19/9/2025, at:
https://acr.ps/1L9BPnQ
[14] Kittichate Visuttijai et al., “Glycogenin Is Dispensable for Glycogen Synthesis in Human Muscle, and Glycogenin Deficiency Causes Polyglucosan Storage,”
Journal of Clinical Endocrinology & Metabolism, vol. 105, no. 2 (2020), pp. 557-566.
[15] “Glycogen Storage Disease (GSD),” Cleveland Clinic, accessed on 19/9/2025, at:
https://acr.ps/1L9BP95
[16] A. Gardiner et al., “Glycogen Storage Disease Type XV: A Case Report,”
Neuromuscular Disorders, vol. 25 (2015), p. 221.
[17] Nuria Mancheño et al., “Cardiac Phenotype in Glycogen Storage Disease Type XV: A Rare Cardiomyopathy to Bear in Mind,”
Revista Española de Cardiología (English Edition), vol. 74, no. 1 (2021), pp. 99-101.
[18] Laura Marr et al., “Mechanism of Glycogen Synthase Inactivation and Interaction with Glycogenin,”
Nature Communications, vol. 13, no. 1, article no. 3372 (2022).
[19] Ying Shen et al., “Molecular Diagnosis of Panel-Based Next-Generation Sequencing Approach and Clinical Symptoms in Patients With Glycogen Storage Disease: A Single Center Retrospective Study,”
Frontiers in Pediatrics, vol. 8, article no. 600446 (2020).
[20] Aitana Almodóvar-Payá et al., “Preclinical Research in Glycogen Storage Diseases: A Comprehensive Review of Current Animal Models,”
International Journal of Molecular Sciences, vol. 21, no. 24, article no. 9621 (2020).
[21] Dwight D. Koeberl et al., “Gene Therapy for Glycogen Storage Diseases,”
Journal of Inherited Metabolic Disease, vol. 47, no. 1 (2024), pp. 93-118.
